






酶
发现及研究史

法国科学家路易·巴斯德
酶的发现来源于人们对发酵机理的逐渐了解。早在18世纪末和19世纪初,人们就认识到食物在胃中被消化, 用植物的提取液可以将淀粉转化为糖,但对于其对应的机理则并不了解。
到了19世纪中叶,法国科学家路易·巴斯德对蔗糖转化为酒精的发酵过程进行了研究,认为在酵母细胞中存在一种活力物质,命名为“酵素”(ferment)。他提出发酵是这种活力物质催化的结果,并认为活力物质只存在于生命体中,细胞破裂就会失去发酵作用。
1878年,德国生理学家威廉·屈内首次提出了 酶 (enzyme)这一概念。随后, 酶 被用于专指胃蛋白酶等一类非活体物质,而 酵素 (ferment)则被用于指由活体细胞产生的催化活性。

德国科学家爱德华·比希纳
这种对酶的错误认识很快得到纠正。1897年,德国科学家爱德华·比希纳开始对不含细胞的酵母提取液进行发酵研究,通过在柏林洪堡大学所做的一系列实验最终证明发酵过程并不需要完整的活细胞存在。 他将其中能够发挥发酵作用的酶命名为发酵酶(zymase)。 这一贡献打开了通向现代酶学与现代生物化学的大门,其本人也因“发现无细胞发酵及相应的生化研究”而获得了1907年的诺贝尔化学奖。在此之后,酶和酵素两个概念合二为一,并依据比希纳的命名方法,酶的发现者们根据其所催化的反应将它们命名。通常酶的英文名称是在催化底物或者反应类型的名字最后加上-ase的后缀,而对应中文命名也采用类似方法,即在名字最后加上“酶”。例如,乳糖酶(lactase)是能够剪切乳糖(lactose)的酶;DNA聚合酶(DNA polymerase)能够催化DNA聚合反应。
人们在认识到酶是一类不依赖于活体细胞的物质后,下一步工作就是鉴定其生化组成成分。许多早期研究者指出,一些蛋白质与酶的催化活性相关;但包括诺贝尔奖得主里夏德·维尔施泰特在内的部分科学家认为酶不是蛋白质,他们辩称那些蛋白质只是酶分子的携带者,蛋白质本身并不具有催化活性。1926年,美国生物化学家詹姆斯·萨姆纳完成了一个决定性的实验。他首次从刀豆得到尿素酶结晶,并证明了尿素酶的蛋白质本质。其后,萨姆纳在1931年在过氧化氢酶的研究中再次证实了酶为蛋白质。约翰·霍华德·诺思罗普和温德尔·梅雷迪思·斯坦利通过对胃蛋白酶、胰蛋白酶和胰凝乳蛋白酶等消化性蛋白酶的研究,最终确认蛋白质可以是酶。以后陆续发现的两千余种酶均证明酶的化学本质是蛋白质。以上三位科学家因此获得1946年度诺贝尔化学奖。
由于蛋白质可以结晶,通过X射线晶体学就可以对酶的三维结构进行研究。第一个获得结构解析的酶分子是溶菌酶,一种在眼泪、唾液和蛋清中含量丰富的酶,其功能是溶解细菌外壳。溶菌酶结构由大卫·菲利浦(David Phillips)所领导的研究组解析,并于1965年发表。 这一成果的发表标志着结构生物学研究的开始,高分辨率的酶三维结构使得对于酶在分子水平上的工作机制的了解成为可能。
1980年代,托马斯·切赫和悉尼·奥尔特曼分别从四膜虫的rRNA前体的加工研究和细菌的核糖核酸酶P复合物的研究中都发现RNA本身具有自我催化作用,并提出了核酶的概念。这是第一次发现蛋白质以外的具有催化活性的生物分子。 1989年,其二人也因此获得诺贝尔化学奖。
结构
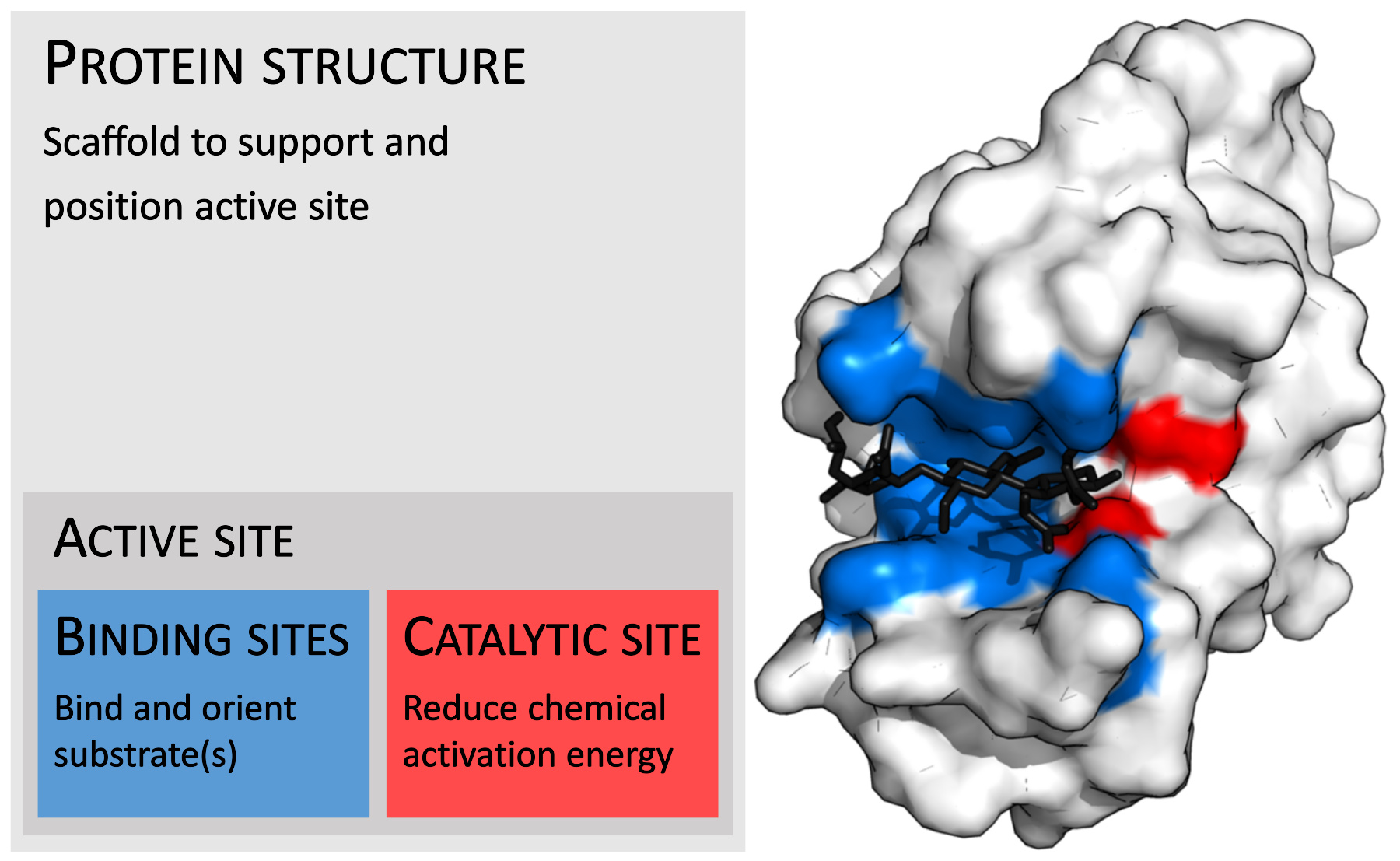
酶结构的组织(以溶菌酶为例)。图中结合位点以蓝色表示,催化位点以红色表示,底物肽聚糖以黑色表示(PDB9LYZ)酶活性最初随温度升高而增加(Q 10为正),直到酶的结构开始去折叠化(即变性)。因而酶促反应的速度在适中的温度条件下是最快的
酶大都是球蛋白,以单体或聚成复合物对反应进行催化。和其他的蛋白质一样,酶的三维结构是通过多肽链折叠形成的。氨基酸的序列(一级结构)能决定蛋白质的三维结构,进而影响酶的催化活性 。尽管结构决定功能是一条具普适性的规则,一种新的酶的活性不能仅仅通过其结构预测 。加热时或与化学变性剂接触时,酶结构会发生去折叠(即变性),原有的结构被打乱,活性也往往随之丧失 。在温度超过正常水平时,酶就会变性。因此,不难推断生活在火山环境(比如热泉)中的细菌的酶具有很强的耐热性。这些酶使高温条件下酶促反应的发生成为可能,在工业上具有很高的利用价值。
酶通常比底物大得多。酶的肽链长度从62个氨基酸残基的 4-草酰巴豆酸异构酶 ( 英语 : 4-Oxalocrotonate tautomerase ) 的单体 到长度超过2,500个氨基酸残基的动物脂肪酸合酶 。酶的结构只有一小部分(大约2-4个氨基酸)是直接与催化相关的。这部分称为催化位点(catalytic site) 。催化位点通常与一个或多个与底物结合的 结合位点 ( 英语 : Binding site ) (binding site)相连。催化位点与结合位点共同组成了酶的活性位点(active site)。酶的其余部分起维持活性位点准确的方向以及动力学特性的作用 。
在一些酶中,催化与任何一个氨基酸都没有关系。这类酶另有与催化辅助因子结合的位点 。一些酶亦可能包含别构位点。小分子与别构位点的结合可使酶发生构象改变,进而使酶的活性降低或升高 。
一些具有生物催化活性的RNA分子称为核酶(ribozyme)。这类分子可能单独发挥催化作用,也可能在与蛋白质结合成复合物的条件下发挥催化作用。最常见的核酶应是核糖体。核糖体是蛋白质以及具有催化活性的RNA的复合物。核糖体的活性位点完全由RNA组成,而蛋白质仅起支架的作用 。
机制
底物结合
酶在催化化学反应前必须要与底物结合。酶具有很强的专一性,通常仅能与寥寥数种底物结合,催化一种或几种反应。专一性通过结合区的形状、电荷、疏水/亲水性与底物互补实现。因此,酶可以用来区分 化学选择性 ( 英语 : chemoselectivity ) 上、区域选择性上、立体专一性上有所不同的结构相似的分子 。
一些与基因组复制与表达相关的酶具有很高的专一性和准确性。一些酶有校对机制。以DNA聚合酶为例,这类酶先催化DNA链的合成,再检查新加上的碱基是否正确 。校对机制确保了酶的极高准确性,哺乳动物的高保真DNA聚合酶在每一亿次反应中才会出一次错误 。RNA聚合酶、氨酰-tRNA合成酶 、核糖体 也有与DNA聚合酶类似的校对机制 。
另外,一些酶表现出 酶乱交 ( 英语 : enzyme promisty ) (enzyme promisty)的现象。这类酶专一性弱,能与一系列生理上相关的底物反应。一些酶偶尔会出现数值不高的副反应活性(即中性进化)。这样的变化可能会成为酶的新功能的进化起点 。
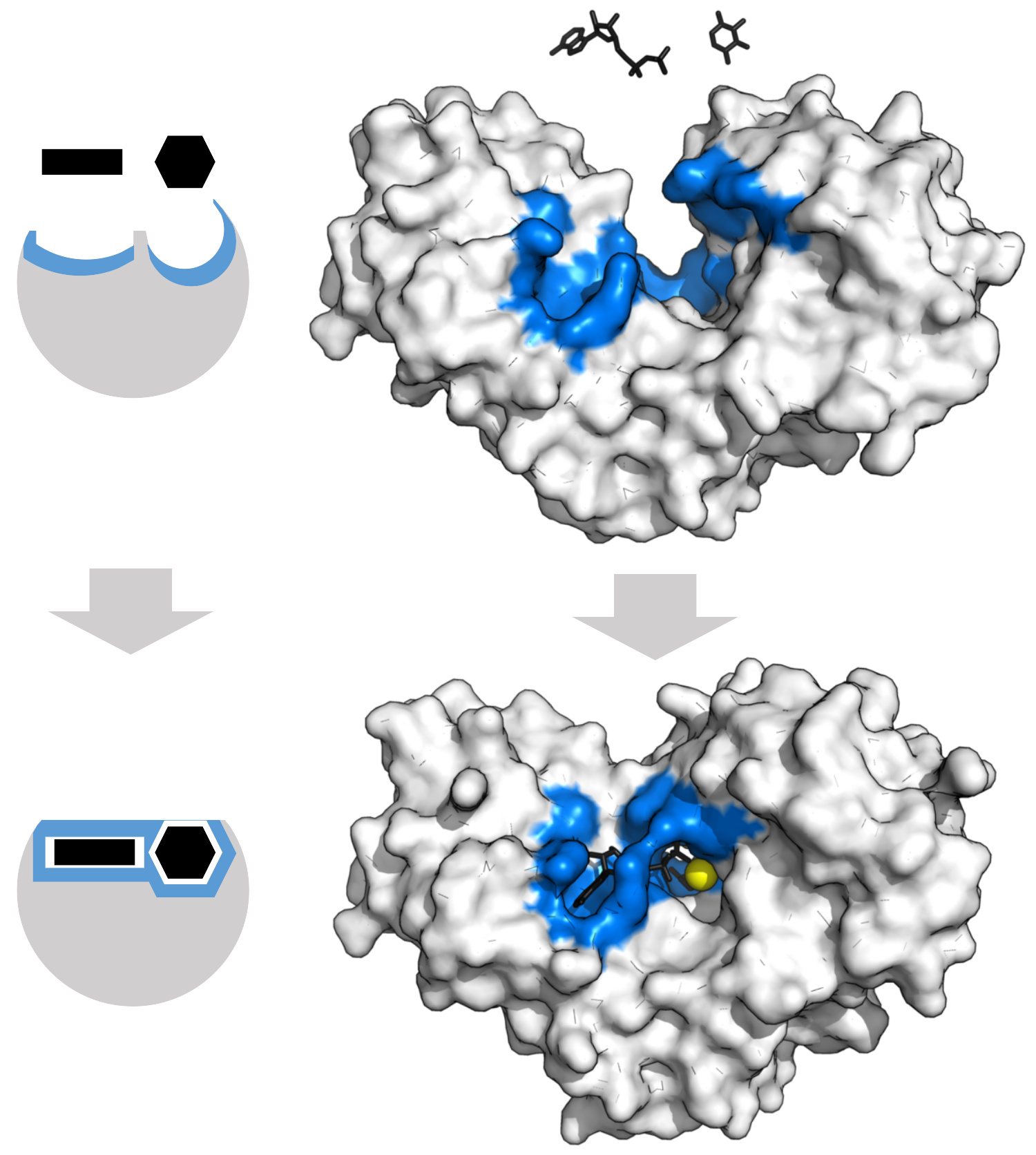
在与底物发生诱导契合后,酶改变形状,生成酶—底物复合物。己糖激酶的诱导契合运动较大,能包覆住底物ATP和木糖。图中结合位点用蓝色表示,底物用黑色表示,Mg 2+辅助因子用黄色表示(PDB2E2N, 2E2Q )
“钥匙和锁”模型
为了解释观察到的酶的特异性,1894年,赫尔曼·埃米尔·费歇尔提出,酶和底物靠着互补的几何形状精准地结合在一起 。这一理论即通常所说的“钥匙和锁”模型 。这一早期的理论解释了酶的专一性,但却没能解释酶的过渡态为何能稳定存在 。
诱导契合模型
1958年, 丹尼尔·科甚兰 ( 英语 : Daniel E. Koshland, Jr. ) 提出了一个对钥匙与锁模型进行修正的理论:酶的结构相对灵活,底物与酶(活性位点)作用时,活性位点会不断改变结构 。底物不是简单地与一个刚性的活性位点结合。组成活性位点的氨基酸侧链的准确有序排布保证酶能执行催化功能。糖苷酶等酶,当底物分子与活性位点结合时,底物分子亦会发生轻微的形状改变 。直到底物与酶发生完全结合,分子形状和电荷排布都最终确定,活性位点都会不断发生结构变化 诱导契合可以通过 结构校对 ( 英语 : Conformational proofreading ) (conformational proofreading)机制在噪音和竞争物存在的条件下增强分子识别的保真度 。
催化作用
酶可以通过多种途径加快化学反应的进行速度。这些途径的机理是降低反应激活能(ΔG ,吉布斯自由能) 。
通过使过度态更加稳定:
提供一条不同的反应途径:
使处于基态的底物稳定性降低:
酶可能会同时使用多种途径催化化学反应。比如胰蛋白酶(trypsin)通过一个催化三联体催化化学反应,借助 氧负离子洞 ( 英语 : oxyanion hole ) (oxyanion hole)改变过渡态的电荷排布,达到增强过渡态稳定性的作用,水解过程的完成则依赖排布有序的水分子。
动力学
酶的结构并不是刚性、恒定不变的。酶的内部结构会发生复杂的动力学变化。酶的结构可能在反应过程中发生变化,单个氨基酸残基、一个 转角 ( 英语 : turn (biochemistry) ) 、一个二级结构单位,乃至整个结构域的位置都可能发生改变。酶内部结构的变化能使差异度不大、且能发生互变的结构之间在热力学平衡状态下发生 构象系综 ( 英语 : conformational ensemble ) (conformational ensemble)。在系综状态下,酶的每一结构状态都可能与酶的功能的某一部分有关。举例来说,二氢叶酸还原酶的不同构象就分别和底物结合、催化、辅助因子释放、产物释放相关 。
别构调节
别构中心是酶当中一个可以让分子和酶结合的区域,别构中心不同于活性中心的。和别构中心结合的分子可以改变酶的构象或是酶的动力学特性,因此影响酶活性中心的反应速率 。因此别构反应可以使酶激活,也可使其不激活。酶和代谢路径上游或是下游代谢物的别构作用会有反馈调整,用路径上的 通量 ( 英语 : Flux (metabolism) ) 来调整酶的活性 。
辅助因子
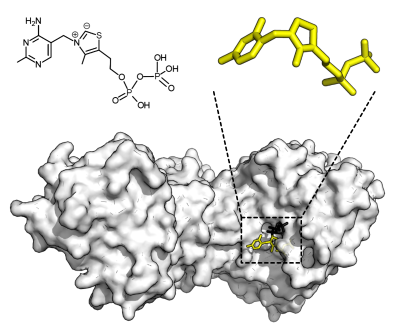
转酮酶 ( 英语 : Transketolase ) (Transketolase)以及硫胺素焦磷酸的结构。黄色部分代表硫胺素焦磷酸,黑色部分代表木酮糖-5-磷酸(PDB4KXV)
一些酶并不需要额外的组分就能就能正常发挥作用,另外一些酶则要在和辅助因子(cofactor)结合后才能显示出活性 。辅助因子可以是无机物(如金属离子、铁硫簇),也可以是有机物(比如黄素和血红素)。有机辅助 因子如果在反应中会与酶分离则为辅酶(coenzyme),如果与酶紧密结合则为辅基(prosthetic groups)。有机的辅基可能与酶发生共价结合(丙酮酸羧化酶与生物素(biotin)之间即发生共价结合) 。
碳酸酐酶 ( 英语 : carbonic anhydrase ) (carbonic anhydrase)即是一类含有辅助因子的酶。 理查德森图 ( 英语 : ribbon diagram ) (ribbon diagram)显示,碳酸酐酶的肽链环绕在一个锌离子(值得注意的是,锌离子亦是活性位点的一部分)周围,并与这个锌离子结合在一起 这些与酶紧密结合的离子或分子通常位于活性位点之中,并且能够参与催化反应 。例如,黄素以及血红素即会参与氧化还原反应 。
需要辅助因子才可以发挥作用的酶,处于未与辅助因子状态时称为“脱辅酶”( apoenzymes 或 apoproteins ),当其与辅助因子结合后则称为“全酶”( holoenzyme )。不过,值得注意的是,名词全酶亦可以指含有多个亚基的酶,如DNA聚合酶。不过,在本文中,全酶是指含有所有发挥活性所需的亚基的酶 。
辅酶
辅酶是一类与酶结合的小分子有机物,辅酶与酶结合的强度因辅酶和酶的种类而异,或强或弱。辅酶能够将化学基团从一个酶转移到另一个酶上 。NADH、NADPH为两种常见的辅酶。核黄素、硫胺、叶酸等维生素类辅酶人体不能合成,需要通过膳食补充。辅酶能携带化学基团,如NAD或NADP能携带氢离子(H )、ATP能携带磷酸基团、辅酶A能携带乙酰基团、叶酸基团能携带甲酰基、次甲基,或甲基、S-腺苷甲硫氨酸能携带甲基 。
辅酶在酶促反应发生后化学结构会发生改变。因此,可以将看作一类普遍存在的特殊底物。许多酶都有与之匹配的辅酶,比如,已知的酶中有大约1000种酶使用NADH作为辅酶(2015年的数据) 。
辅酶通常能不断再生,浓度能始终维持在一个恒定不变的水平上。举例来说,NADPH能通过磷酸戊糖途径再生,S-腺苷甲硫氨酸能通过甲硫氨酸腺苷转移酶催化的反应生成。持续不断的再生意味着总量不多的辅酶也能以很快的速度消耗。举例来说,人类每天会更新重量与自身体重相等的ATP 。
热力学
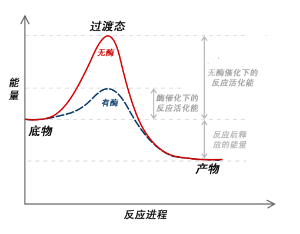
有酶或无酶催化反应体系中反应进程与能量关系图示。可以看出,当反应没有酶的催化时,底物通常需要获得较高的激活能才能到达过渡态,然后才能生成产物;而当反应体系中有酶催化时,通过酶对过渡态的稳定作用,降低了达到过渡态所需能量,从而降低了整个反应所需的能量。
与其他催化剂一样,酶并不改变反应的平衡常数,而是通过降低反应的激活能来加快反应速率(见右图)。通常情况下,反应在酶存在或不存在的两种条件下,其反应方向是相同的,只是前者的反应速度更快一些。但必须指出的是,在酶不存在的情况下,底物可以通过其他不受催化的“自由”反应生成不同的产物,原因是这些不同产物的形成速度更快。
酶可以连接两个或多个反应,因此可以用一个热力学上更容易发生的反应去“驱动”另一个热力学上不容易发生的反应。例如,细胞常常通过ATP被酶水解所产生的能量来驱动其他化学反应。
酶可以同等地催化正向反应和逆向反应,而并不改变反应自身的化学平衡。例如,碳酸酐酶可以催化如下两个互逆反应,催化哪一种反应则是依赖于反应物浓度。
反应式中“*”表示“碳酸酐酶”
当然,如果反应平衡极大地趋向于某一方向,比如释放高能量的反应,而逆反应不可能有效的发生,则此时酶实际上只催化热力学上允许的方向,而不催化其逆反应。
动力学
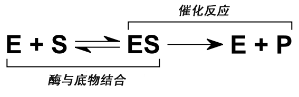
单一底物的酶催化反应机理:酶(表示为“E”)结合底物(表示为“S”),并通过催化反应生成产物(表示为“P”)。
酶动力学是研究酶结合底物能力和催化反应速率的科学。研究者通过酶反应分析法(enzyme assay)来获得用于酶动力学分析的反应速率数据。
1902年,维克多·亨利提出了酶动力学的定量理论; 随后该理论得到他人证实并扩展为米氏方程。 亨利最大贡献在于其首次提出酶催化反应由两步组成:首先,底物可逆地结合到酶上,形成酶-底物复合物;然后,酶完成对对应化学反应的催化,并释放生成的产物(见左图)。
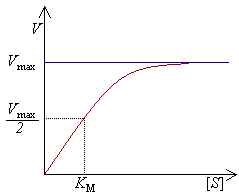
酶初始反应速率(表示为“ V ”)与底物浓度(表示为“[S]”)的关系曲线。随着底物浓度不断提高,酶的反应速率也趋向于最大反应速率(表示为“ V max ”)。
酶可以在一秒钟内催化数百万个反应。例如,乳清酸核苷5"-磷酸脱羧酶所催化的反应在无酶情况下,需要七千八百万年才能将一半的底物转化为产物;而同样的反应过程,如果加入这种脱羧酶,则需要的时间只有25毫秒。 酶催化速率依赖于反应条件和底物浓度。如果反应条件中存在能够将蛋白解链的因素,如高温、极端的pH和高的盐浓度,都会破坏酶的活性;而提高反应体系中的底物浓度则会增加酶的活性。在酶浓度固定的情况下,随着底物浓度的不断升高,酶催化的反应速率也不断加快并趋向于最大反应速率( V max ,见右图的饱和曲线)。出现这种现象的原因是,当反应体系中底物的浓度升高,越来越多自由状态下的酶分子结合底物形成酶-底物复合物;当所有酶分子的活性位点都被底物饱和结合,即所有酶分子形成酶-底物复合物时,催化的反应速率达到最大。当然, V max 并不是酶唯一的动力学常数,要达到一定反应速率所需的底物浓度也是一个重要的动力学指标。这一动力学指标即米氏常数( K m ),指的是达到 V max 值一半的反应速率所需的底物浓度(见右图)。对于特定的底物,每一种酶都有其特征 K m 值,表示底物与酶之间的结合强度( K m 值越低,结合越牢固,亲和力越高)。另一个重要的动力学指标是 k cat ,定义为一个酶活性位点在一秒钟内催化底物的数量,用于表示酶催化特定底物的能力。
酶的催化效率可以用 k cat / K m 来衡量。这一表示式又被称为特异性常数,其包含了催化反应中所有步骤的反应常数。由于特异性常数同时反映了酶对底物的亲和力和催化能力,因此可以用于比较不同酶对于特定底物的 催化效率或同一种酶对于不同底物的催化效率。特异性常数的理论最大值,又称为扩散极限,约为10 至10 M s ;此时,酶与底物的每一次碰撞都会导致底物被催化,因此产物的生成速率不再为反应速率所主导,而分子的扩散速率起到了决定性作用。酶的这种特性被称为“催化完美性”或“动力学完美性”。相关的酶的例子有磷酸丙糖异构酶、碳酸酐酶、乙酰胆碱酯酶、过氧化氢酶、延胡索酸酶、β-内酰胺酶和超氧化物歧化酶。
米氏方程是基于质量作用定律而确立的,而该定律则基于自由扩散和热动力学驱动的碰撞这些假定。然而,由于酶/底物/产物的高浓度和相分离或者一维/二维分子运动,许多生化或细胞进程明显偏离质量作用定律的假定。 在这些情况下,可以应用分形米氏方程。
存在一些酶,它们的催化产物动力学速率甚至高于分子扩散速率,这种现象无法用目前公认的理论来解释。有多种理论模型被提出来解释这类现象。其中,部分情况可以用酶对底物的附加效应来解释,即一些酶被认为可以通过双偶极电场来捕捉底物以及将底物以正确方位摆放到催化活性位点。另一种理论模型引入了基于量子理论的穿隧效应,即质子或电子可以穿过激活能垒(就如同穿过隧道一般),但关于穿隧效应还有较多争议。 有报道发现色胺中质子存在量子穿隧效应。 因此,有研究者相信在酶催化中也存在着穿隧效应,可以直接穿过反应能垒,而不是像传统理论模型的方式通过降低能垒达到催化效果。有相关的实验报道提出在一种醇脱氢酶的催化反应中存在穿隧效应, 但穿隧效应是否在酶催化反应中普遍存在并未有定论。
抑制作用
酶的催化活性可以被多种抑制剂所降低。
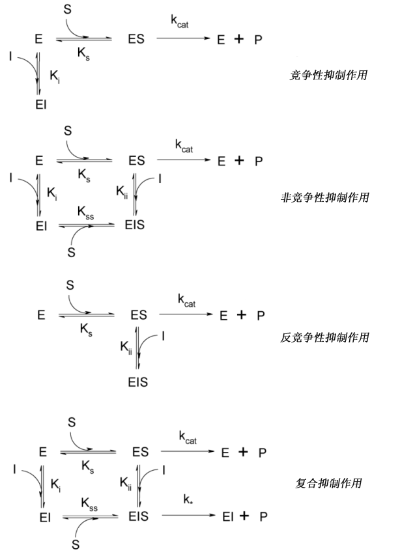
不同的抑制类型。分类参考自 。图中,“E”表示酶;“I”表示抑制剂;“S”表示底物;“P”表示产物。
可逆抑制作用
可逆抑制作用的类型有多种,它们的共同特点在于抑制剂对酶活性的抑制反应具有可逆性。
竞争性抑制作用
抑制剂与底物竞争结合酶的活性位点(抑制剂和底物不能同时结合到活性位点),也就意味着它们不能同时结合到酶上。 对于竞争性抑制作用,催化反应的最大反应速率值没有变,但是需要更高的底物浓度,反映在表观 K m 值的增加。
非竞争性抑制作用
非竞争性抑制抑制剂可以与底物同时结合到酶上,即抑制剂不结合到活性位点。酶-抑制剂复合物(EI)或酶-抑制剂-底物复合物(EIS)都没有催化活性。与竞争性抑制作用相比,非竞争性抑制作用不能通过提高底物浓度来达到所需反应速度,即表观最大反应速率 V max 的值变小;而同时,由于抑制剂不影响底物与酶的结合,因此 K m 值保持不变。
反竞争性抑制作用
反竞争性抑制作用比较少见:抑制剂不能与处于自由状态下的酶结合,而只能和酶-底物复合物(ES)结合,在酶反应动力学上表现为 V max 和 K m 值都变小。这种抑制作用可能发生在多亚基酶中。
复合抑制作用
这种抑制作用与非竞争性抑制作用比较相似,区别在于EIS复合物残留有部分酶的活性。在许多生物体中,这类抑制剂可以作为负反馈机制的组成部分。若一个酶体系生产了过多的产物,那么产物就会抑制合成该产物的酶体系中第一个酶的活性,这就可以保证一旦合成足够多的产物后,该产物的合成速率会下降或停止。受这种抑制作用调控的酶通常为多亚基酶,并具有与调控产物结合的别构结合位点。这种抑制作用的反应速率与底物浓度的关系图不再是双曲线形而是S形。
不可逆抑制作用
不可逆抑制剂可以与酶结合形成共价连接,而其他抑制作用中酶与抑制剂之间都是非共价结合。这种抑制作用是不可逆的,酶一旦被抑制后就无法再恢复活性状态。这类抑制剂包括二氟甲基鸟氨酸(一种可用于治疗寄生虫导致的昏睡症的药物 )、苯甲基磺酰氟(PMSF)、青霉素和阿司匹林。这些药物都是与酶活性位点结合并被激活,然后与活性位点处的一个或多个氨基酸残基发生不可逆的反应形成共价连接。
抑制剂的用途
酶抑制剂常被用作药物,同样也可以被作为毒药使用。而药物和毒药之间的差别通常非常小,大多数的药物都有一定程度的毒性,正如帕拉塞尔苏斯所言:“所有东西都有毒,没有什么是无毒的”(“In all things there is a poison, and there is nothing without a poison”)。 相同的,抗生素和其他抗感染药物只是特异性地对病原体而不是对宿主有毒性。
一个获得广泛应用的抑制剂药物是阿司匹林,它可以抑制环加氧酶的活性,而环加氧酶可以生产炎症反应信使前列腺素,因此,阿司匹林可以起到抑制疼痛与炎症的作用。而剧毒毒药氰化物可以通过结合细胞色素氧化酶位点处的铜和铁原子不可逆地抑制酶活性,从而抑制细胞的呼吸作用。
活性控制
细胞内有五种控制酶催化活性的机制:
根据外界环境的变化,细胞可以增强或减弱酶的生产(即酶相关基因的转录和翻译)。这属于一种基因调控,被称为酶的诱导和抑制。例如,当环境现如青霉素这样的抗生素时,部分细菌可以对抗生素产生抗性,其原因就在于细菌体内的β-半乳糖苷酶被诱导而大量生产,这种酶可以水解青霉素分子上关键的β-乳胺环。另一个例子是在人体肝脏中存在一类酶对于药物代谢非常重要的酶,细胞色素P450氧化酶;对这一类酶的诱导或抑制,会导致 药物相互作用 ( 英语 : drug interaction ) 。
通过将特定的酶分隔在特定的细胞组分中,细胞可以完成不同的代谢途径。例如,脂肪酸的合成是由细胞溶质、内质网和高尔基体中的一系列酶所完成,而脂肪酸的降解(以提供能量)是在线粒体中由另一系列酶通过β-氧化来完成。
酶可以被抑制剂与激活剂所调控。例如,一个代谢途径中的终产物常常是这一途径中第一个酶的抑制剂,从而调控这一代谢途径的产物量。这种调控机制被称为负反馈机制,因为终产物的合成量是受其自身浓度调控。负反馈机制可以根据细胞的需要,有效地调节中间代谢物的合成速率,从而使细胞的能量和物质的分配更为高效,并防止多余产物的合成。控制酶的作用,可以在生物体内维持一个稳定的内部环境(即体内平衡)。
翻译后修饰也可以调控酶的活性。这些修饰包括磷酸化、肉豆蔻酸化和糖基化。例如,细胞接受胰岛素信号后,对包括糖原合酶在内的多个酶进行磷酸化,帮助控制糖原的合成或降解,使得细胞可以对血糖的变化产生反应。 另一个翻译后修饰的例子是多肽链的剪切。胰凝乳蛋白酶,一种消化性蛋白酶,是产生于胰脏中的无活性的胰凝乳蛋白酶原,这一蛋白通过运输到达胃后才被激活。这种方式有效地防止了胰凝乳蛋白酶在进入肠之前消化胰脏或其他组织。这种无活性的酶的前体被命名为酶原。
还有一些酶可以通过定位到不同环境后而被激活,比如从还原态的环境(细胞质)到氧化态环境(细胞周质空间),从高pH环境到低pH环境等。流感病毒的红血细胞凝集素蛋白就是一个例子:当它接触到宿主细胞囊泡的酸性环境时,它的构象立刻发生变化,导致其获得激活。
相关疾病
苯丙氨酸羟化酶. 由PDB 1KW0生成。
酶的活性必须严格控制以维持体内平衡,对于能够影响一个关键酶的功能的任何基因缺陷(如突变导致活性变化,过量表达、过低表达或删除突变)都可能导致遗传性疾病发生。许多事实显示,一种致命疾病的病因可以只是由于人体中的数千种酶中的一种发生功能故障。
苯丙酮尿症:此种病症是典型的酶相关病例之一。病因是苯丙氨酸羟化酶(其功能是催化苯丙氨酸降解过程中的第一步)上一个氨基酸位点发生了突变,导致体内苯丙氨酸和相关产物的水平过高,如果没有得到合适的治疗,会进一步导致智能障碍。
卟啉病:该病是由于血红素生物合成途径中特定酶的酶活性过低(基因突变或其他原因导致),使得中间产物卟啉的产生和排泄异常,在一定诱因(如阳光照射)下,可导致皮肤或其他组织器官发生病变。
当生殖细胞中编码DNA修复相关酶的基因发生突变,其结果会导致遗传性癌症综合病征,如着色性干皮症。 DNA修复酶的缺陷导致人体丧失修复突变基因的能力。发生的突变不断积累,最终使得患者有多种癌症发生。
酶的口服给药,可用于治疗多种疾病(如胰腺功能不全和乳糖不耐受症)。由于酶作为蛋白质可能在消化道环境中失活或被降解,因此一种非侵入性的成像方法被开发用于监测作为药物的酶在消化道中的活性变化。
命名规则
酶的命名是衍生自其底物或是要催化的化学反应,在字尾会加上 -ase 。例如乳糖酶(lactase)、醇脱氢酶(alcohol dehydrogenase)及DNA聚合酶(DNA polymerase)。但有些化学反应可以由几种不同的酶催化,这些酶称为同工酶 ,而上述的命名法无法处理同工酶的情形。
国际生物化学与分子生物盟 ( 英语 : International Union of Biochemistry and Molecular Biology ) 提出了酶的 命名法 ( 英语 : nomenclature ) ,也就是EC编号。每一个酶用一个四位数的数字表示,前面再加上"EC"。第一位数字是酶依酶促反应的机制来分类 。
依照第一位数字,可以分为以下六类:
EC 1氧化还原酶:催化氧化还原反应的酶类,例如乳酸脱氢酶、琥珀酸脱氢酶、细胞色素氧化酶、过氧化氢酶、过氧化物酶等。
EC 2转移酶:转移官能团(例如甲基或是磷酸基团)的酶类,例如 甲基转移酶 ( 英语 : Methyltransferase ) 、氨基转移酶、己糖激酶、 磷酸化酶 ( 英语 : Phosphorylase ) 等。
EC 3水解酶:催化底物发生水解反应的酶类,例如淀粉酶、蛋白酶、脂肪酶、磷酸酶等。
EC 4裂合酶:用氧化及水解反应以外的方式移去基团的酶类,例如 碳酸酐酶 ( 英语 : Carbonic anhydrase ) 、醛缩酶、柠檬酸合酶等。
EC 5异构酶:催化分子同分异构反应的酶类,例如磷酸丙糖异构酶、 消旋酶 ( 英语 : Racemase ) 等。
EC 6连接酶:用共价键结合二个分子的酶类,例如谷氨酰胺合成酶、丙酮酸羧化酶等。。
上述分类会再依底物、生成物及化学结构来分类。用四位数字可以完整的描述一个酶。例如,己糖激酶(EC 2.7.1.1)是转移酶(EC 2),会将磷酸基团(EC 7)加到六碳糖中,是一个含有醇基的分子(EC 2.7.1) 。
各行业的应用
酶因为能高效催化特定反应,已在化工等行业得到广泛应用。总的来说,酶的应用因为它们能催化的反应数目少、在有机溶剂中以及高温环境下不稳定而受到限制。因此,酶工程这一热门学科应运而生。酶工程旨在借助合理的设计或体外进化的方法研发具有新特性的酶 。目前,酶工程学已取得了一定成果,研究人员甚至已“从头”(即不以任何自然界中的酶为模板)设计出了一些能催化在自然界中不能发生的反应的酶 。
参见
酶列表
酶数据库
拓展阅读
免责声明:以上内容版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。感谢每一位辛勤著写的作者,感谢每一位的分享。

相关资料





- 有价值
- 一般般
- 没价值















推荐阅读

关于我们

APP下载


