






化学反应
类别
有几种主要化学反应如下所示:
化合反应( A + B → → --> C {\displaystyle {\rm {A+B\rightarrow C\,}}} ):由两种或两种以上物质生成另一种物质的反应
分解反应( A → → --> B + C {\displaystyle {\rm {A\rightarrow B+C\,}}} ):由一种反应物生成两种或两种以上其他物质的反应
无机反应:指以无机化合物为反应物的各种反应。
有机反应:指有机化合物为反应物的各种反应。
酸碱反应:指酸与碱发生作用生成“酸碱加合物”的过程。这里的酸、碱可以有不同的定义:
氧化还原反应:指涉及电子转移的反应(如:单取代反应和燃烧反应)。
燃烧反应:指底物与氧化剂(比如氧气)发生的剧烈氧还反应,反应中通常伴有发光、放热等现象。(本质上属于氧化还原反应。)
当然还有更多复杂的情形,但仍可逐步简单化而视为上述反应类别的连续反应。化学反应的变化多端难以建立简单的分类标准。更多的例子参见化学反应列表。
反应与能量
能量净改变
根据热力学第二定律,任何等温等压封闭系统倾向降低吉布斯能。在没有外力的影响下,任何反应混合物也是如此。比方,对系统中焓的分析可以得到合乎反应混合物的热力学计算。反应中焓的计算方式采用标准反应焓以及反应热加成性定律(赫士定律/盖斯定律)。
以甲烷在氧中的燃烧反应为例: C H 4 + 2 O 2 → → --> C O 2 + 2 H 2 O {\displaystyle {\rm {CH_{4}+2O_{2}\rightarrow CO_{2}+2H_{2}O\,}}}
能量计算须打断反应左侧和右侧的所有键结取得能量数据,才能计算反应物和生成物的能量差。以ΔH表示能量差,Δ(Delta)表示差异,H则为焓等于固定压力下的热传导能量。ΔH的单位为千焦耳或千卡。
放热反应:如果ΔH计算为负值,则反应能量通常会释放出来形成热,即为放热反应。放热反应会自发性产生。比如日常所知的燃烧反应就是在空气中燃烧瓦斯产生热量。
吸热反应:也有反应是正值的ΔH。如果 ΔH是正值,完成反应须吸收能量。即所谓的吸热反应。
反应自发性
上述法则“放热反应会自发性产生”通常是事实。但在某些状况下却不是如此,某些吸热反应,如硝酸铵溶于水的过程,虽是从环境吸收能量的反应,但仍然是自发的。这是因为反应的自发性不仅取决于反应热(焓)的变化,也与另一个热力学函数熵有关。熵是表明微观状态的混乱度的函数。对于同一反应物而言,产物中粒子可以采取的微观状态数越多,混乱程度越大,那么反应的熵变也相应地增加。
吉布斯能——封闭系统在等温等压条件下向环境可能做的最大有用功,是用于判断反应自发性的一个状态函数。它综合考虑了焓与熵对反应自发性的贡献,当它为负值时,反应自发;当它为正值时,反应非自发;当它为零时,反应达到平衡态。其基本方程式如下:
通过推导,还可以将吉布斯能与宇宙的熵变(系统+环境)联系起来,得到下面的方程: Δ Δ --> S u n i v = − − --> Δ Δ --> G T {\displaystyle {\rm {\Delta S_{univ}=-{\frac {\Delta G}{T}}\,}}} 。根据热力学第二定律,宇宙的熵总是倾向于增加,ΔSuniv大于零,因此ΔG小于零时,反应是自发的,也得到与上面相同的结论。
反应动力学
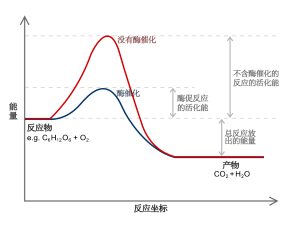
图示为一个反应坐标,当中位于能量最高点的均为过渡态。红色线表示了没有加入催化剂(酶)因此具有较高的活化能,蓝色线是加了催化剂后的情况。
热力学可回答“反应可能发生吗?”,另一个问题“反应多快?”却完全没有回答。这是因为热力学或者热力学平衡试着要了解的是反应混合物初始和结束状态。因此无法指出反应发生时的过程。这个领域属于反应动力学的范畴。
右图称为反应坐标图,是一个常用于表明化学反应中能量变化过程的图。横坐标表示反应进程,显示的是反应物质经由反应物、过渡态,转化为产物的过程;纵坐标表示反应物质的相对能量。反应进程中能量的最高点与反应物的能量之差称为活化能,是为了使反应发生,反应物应具有的最低能量。反应需要越过一个能量高的过渡态,如同从一个谷地爬山到达另一个谷地一样。
分子动力学中,为了解释化学反应的发生,人们提出了很多理论。过渡态理论认为在基元反应中,反应物分子在合适的碰撞取向上相互碰撞后,价电子云相互穿透,形成活化复合物。该络合物也就是右图中能量最高的一点,称为过渡态。这时反应物原有的化学键部分断裂,新的化学键部分地形成。如果反应完成,则旧键破裂,新键形成,转化为产物分子。另一个常见的理论是碰撞理论,它认为,反应物分子间的相互碰撞是反应进行的必要条件,并因此提出有效碰撞、活化分子、频率因子、取向因子、能量因子等概念。碰撞理论较为简单直观,对于简单的双分子反应比较适用,如果反应过于复杂,那么碰撞理论的结果往往与事实有偏差。
很多看上去很简单的反应往往不是一步完成的。它们被称为非基元反应,其中的每一步反应称为基元反应。分步的化学反应往往会产生一些中间物质,称为反应中间体,它们由反应物反应得到,若进一步反应则转化为产物。而且,各基元反应的反应速率往往也不相同,较慢的反应卡住了反应进程,控制了反应的速率方程(速率与反应物浓度的关系),称为反应的速率控制步骤,简称“决速步”。
反应速率
化学反应的反应速率是相关受质浓度随时间改变的的测量。它总是正值。对于反应 a A + b B → → --> p P + q Q {\displaystyle {\rm {aA+bB\rightarrow pP+qQ\,}}} ,反应速率的表达式为:
反应速率的分析有许多重要应用,像是化学工程学或化学平衡研究。反应速率受到下列因素的影响:
反应物浓度:如果增加通常将使反应加速。
活化能:定义为反应启始或自然发生所需的能量。愈高的活化能表示反应愈难以启始,反应速率也因此愈慢。
接触的表面积:表面积越大,反应物之间的碰撞次数越多。
压强:压强越大,反应物之间的碰撞越频繁,因此有效碰撞数也会相应增加。
反应温度:温度提升将加速反应,因为愈高的温度表示有愈多的能量,使反应容易发生。
催化剂:催化剂是一种透过降低活化能提升反应速率的物质。而且催化剂在反应过程中没有净消耗,所以可以重复作用。
反应速率与参与反应的物质浓度有关。物质浓度则可透过质量作用定律定量。
可逆反应
每个化学反应理论上均是可逆反应。正反应中定义物质从反应物转换成产物。逆反应刖相反,产物转换成反应物。
化学平衡指正反应速率和逆反应速率达到相等的状态,因此反应物和产物均会存在。然而,平衡态的反应方向可透过改变反应状态改变,譬如温度或压力。勒沙特烈原理在此用来预测是产物或反应物形成。
平衡常数可用于表明反应达到平衡时反应的进行程度。对于反应 α α --> A + β β --> B . . . ⇌ ⇌ --> σ σ --> S + τ τ --> T . . . {\displaystyle \alpha A+\beta B...\rightleftharpoons \sigma S+\tau T...} ,其平衡常数可以写为 K = { S } σ σ --> { T } τ τ --> . . . { A } α α --> { B } β β --> . . . {\displaystyle K={\frac {{\{S\}}^{\sigma }{\{T\}}^{\tau }...}{{\{A\}}^{\alpha }{\{B\}}^{\beta }...}}} 。括弧代表该物质的活度,粗略计算时可以看做相应的浓度。平衡常数越大,表示达到平衡时产物越多,反应物越少,因此反应趋势或彻底性更大。平衡常数与吉布斯能之间存在如下关系: Δ Δ --> r G m ⊖ ⊖ --> = − − --> R T ln --> K {\displaystyle \ {\Delta }_{r}G_{m}^{\ominus }=-RT\ln K} ,根据上面对反应自发性的讨论,也可以类推出平衡常数与自发性之间的关系。K > 1时反应自发。
虽然所有的反应在一些范围内均可视为微观可逆,部分反应仍可归类为不可逆反应。“不可逆反应”指得是“比较彻底的反应”,意思是几乎所有的反应物均形成产物。反应平衡常数非常大,在达到平衡时,反应物的浓度可以忽略不计。对于究竟多彻底的反应才能被称为不可逆反应,各处的评判标准不同,一般认为平衡常数大于10的反应算是比较彻底的反应。
参见
化学反应列表
化学方程式
热力学、热化学
化学动力学、化学平衡
化学变化、物理变化
免责声明:以上内容版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。感谢每一位辛勤著写的作者,感谢每一位的分享。

相关资料
展开


- 有价值
- 一般般
- 没价值
















关于我们

APP下载


