






过渡态理论
理论
过渡状态理论的基础思想如下:
通过研究建立在势能平面上的马鞍点处的活化络合物来研究反应速率。而该活化络合物是怎样形成的细节并不重要。
活化络合物与反应物分子处于特定的平衡(准平衡)。
活化络合物可以转化成不同的产物,而,这种转化速率可以用动力学理论计算。
发展
在TST的发展过程中,主要采取了如下三种方法。
热力学处理
在1884年,雅各布斯·亨里克斯·范托夫提出了描述温度对可逆反应平衡常数的依赖关系的范特霍夫方程:
其中,ΔU是内能的变化量,K是反应的平衡常数,R是普适气体常数,而T是热力学温度。基于实验工作,1889年,斯凡特·阿瑞尼士提出了一个相似的对于反应常数的表达式:
这个表达式的积分形式,引出了阿伦尼乌斯方程:
A叫做频率因子(现在叫指前因子),E被看作为活化能。到20世纪初,许多人接受了阿伦尼乌斯方程,但是,A 和 E的物理学解释仍还比较模糊。这就导致了许多化学动力学领域的研究人员提出了关于化学反应如何进行的不同的理论,主要试图将A 和 E与直接负责化学反应的分子热运动相关联。
1910年,热内·马塞林(英语:Rene Marcelin)(Rene Marcelin)引入了标准吉布斯活化能。他的方程可写成:
在大约与热内·马塞林同时研究他这个公式的同时,三位荷兰化学家Philip Abraham Kohnstamm(英语:Philip Abraham Kohnstamm)、Frans Eppo Cornelis Scheffer(英语:Frans Eppo Cornelis Scheffer)、和Wiedold Frans Brandsma(英语:Wiedold Frans Brandsma)首次提出了标准活化熵和标准活化焓的概念。他们提出的速率常数方程如下:
然而,常数的性质仍不清楚。
动力学理论处理
上世纪初,Max Trautz(英语:Max Trautz)和William Lewis(英语:William Lewis (chemist))基于气体的动力学理论,用碰撞理论研究反应速率。碰撞理论将反应中的分子处理成相互碰撞的硬质小球;该理论忽视熵变。
Lewis用他的这种处理跟踪反应并和实验结果取得了很好的一致性。
2HI → H2 + I2
然而,此后当这种处理方法用于其他反应时,在理论和实验结果上出现了差异性。
统计热力学处理
在TST理论的发展过程中,统计力学扮演了非常重要的角色。然而,如果考虑到在19世纪中叶,麦克斯韦、玻尔兹曼和Leopold Pfaundler(英语:Leopold Pfaundler)就已经发表了几篇用分子动力学和分子速度的统计分布的概念讨论反应平衡和速率的几篇论文这一事实的话,统计力学在发展TST方面的应用过程是非常慢的。
直到1912年当法国化学家A. Berthoud才使用麦克斯韦-玻尔兹曼分布定律,获得了对速度常数的表达:
其中 a 和 b 是与能量项相关的常数。
两年以后,Marcelin通过把化学反应过程处理成一个点在项空间的运动对TST理论做出了关键性的贡献。然后,他使用吉布斯的统计力学过程取得了一个相似于他早年已经从热力学的考虑取得的方程相似的表达。
1915年,英国物理学家James Rice(英语:James Rice)对TST做出了另一关键性的贡献。基于其统计分析他得出结论反应速率常数正比于“临界增量”。 Tolman进一步发展了他的观点。1919年,奥地利物理学家Karl Ferdinand Herzfeld(英语:Karl Ferdinand Herzfeld)将统计力学用于平衡常数并将动力学理论用于可逆反应的速率常数,k-1对于双原子分子的可逆分解:
他取得了对于正反应的速率常数的方程:
其中E⊖ ⊖ -->{\displaystyle \textstyle E^{\ominus }绝对零度对零度时反应的离解能,kB是波尔兹曼常数,h是普朗克常数,T是热力学温度,υ是键的振动频率。这个表述是非常重要的,因为是kBT/h因子第一次出现在速率方程中,这个因子是TST至关重要的部分。
1920年,美国化学家Richard Chase Tolman(英语:Richard Chase Tolman)进一步发展了Rice关于临界增量的概念。他得出结论一个反应的临界增量(现在叫做活化能)等于所有经历反应的分子的平均能量减去所有反应物分子的平均能量。
势能面
在TST的发展过程中,势能面的概念非常重要。这个理论的基础是由Marcelin奠定的。他提出的理论认为:化学反应的进程应该被描述为在具有原子动量和距离两个坐标的势能面上的一个点。
1931年,亨利·艾林和迈克尔·波拉尼构造了下列反应的势能面。这个面是一个基于量子力学原理以及振动频率和离解能的实验数据的三维面。
H + H2 → H2 + H
在艾林 和 波拉尼做出这一贡献后的一年,H. Pelzer and Eugene Wigner通过跟踪一个势能面上的反应过程做出了一项重大的贡献。他们的工作的重要性在于是他们首次讨论了势能面上的“山坳”或“马鞍”点问题。他们的结论是反应速率由体系通过山坳的运动来决定。
艾林方程的推导
亨利·艾林、迈克尔·波拉尼和Evans所加入的唯一重要特点是活化络合物与反应物处于准平衡的概念。反应速率正比于这些络合物的浓度乘以它们转化成产物的频率(kBT/h)。
准平衡态假设
应该注意的是准平衡态不同于经典的化学平衡,但是可以用同样的热力学处理方法描述。考虑如下反应,
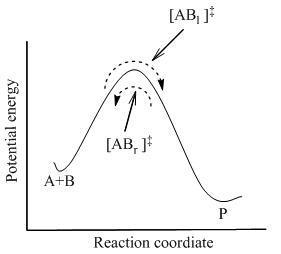
Figure 2: Potential energy diagram
这里体系中所有物种,包括活化络合物,间达到了完全的平衡,[AB]。使用统计热力学,[AB]的浓度可计算成以A和B的浓度所表达的浓度。
TST假设甚至在反应物和产物未相互达到平衡的时候,活化络合物与反应物仍处于准平衡态。如图2所示,在任何时刻,都将有少量的活化络合物。其中一些是是刚刚由反应物分子变来的,表达成[ABl](由于它们正在从左往右运动)。另外的那些是刚刚从产物变来的,[ABr]。由于体系处于完全平衡态,[ABl] 的浓度与[ABr]相等,以至每种的浓度都等于活化络合物的总浓度的一半:
如果产物分子突然间从反应体系中拿掉,这些开始作为产物([ABr] )的活化络合物的流动将停止;然而,仍然有一个从左向右的流动。因而,假设为从左到右的流动速率不受产物移掉的影响;换句话说,两个方向的流动假设是相互独立的。
在TST中,重要的是要离解到当说到活化络合物与反应物平衡,仅仅是指那些刚刚从反应物分子变来的活化络合物([ABl] )。
对于该准平衡态的平衡常数K可以写成:
这样,过渡态AB的浓度是
因而,对产物产生的速率方程为
这里速率常数k表示为
k 直接正比于对应于活化络合物向产物转化的振动模型的振动频率;这个振动模式的频率为ν。不是每一次振动都必然导致产物的生成,因而,需要一个叫做传输系数(transmission coefficient)的比例常数κ的引入,来考虑这种效应。所以,k可以被重写成
对于平衡常数K 统计力学于温度的依赖关系的表述:
其中
将新的对k and K的表述结合起来,可以写出一个新的速率常数的表达式,如下:
由于ΔG = ΔH –TΔS,可以将速率常数的表达式扩充,得到艾林方程:
TST的速率常数表达式可以用于计算ΔG、ΔH、ΔS和甚至ΔV(用速率数据表达的(活化体积)。
过渡态理论的局限性
总的说来,TST已经给研究者提供了一个了解化学反应是如何发生的的概念基础。尽管该理论被广泛地接收,但它依然存在着局限性。 例如,该理论假设过渡结构走向势能面的下坡,它将导致一种产物分子(或一组产物),然而,在一些反应中,过渡态可以以某种方式跨过势能面,导致用过渡态理论一种未曾预料的产物的选择。(这种反应的一个例子是由Anslyn and Dougherty提供的重氮二环戊烷类的热分解反应)。
过渡态理论也假定原子核的行为符合经典力学理论的假设。即假设只有能量足够的原子或分子碰撞才能形成过渡态,否则,不会发反应。然而,根据量子力学,对于任何有限能量的能垒,都有一些粒子仍然能够穿过能垒的可能性。结合到化学反应这就意味着甚至那些不具备足够能量穿过势能面的分子间的碰撞也有机会发生反应。 当这对于高活化能的反应这种效果常常被人们忽略的同时,它对相对低能垒的反应则成为一种重要的现象,因为穿过活化能垒的几率随着活化能的降低而升高。
过渡态理论不适用于高温反应。该理论假设反应体系通过势能面上最低的马鞍点。回想一下,最高的马鞍点叫做过渡态。当该描述与相对低温发生的反应一直时,然而,在高温下分子普遍具有较高能量的振动模式:它们的运动成为更复杂碰撞可能导致远离由过渡态能量所预期的过渡态。这个对过渡态理论的偏离甚至在双原子氢和氢自由基的交换反应中可以观察到。
由于存在这些局限性,有人建议了几种对过渡态理论的修正。下面将做一个简要的讨论。
广义的过渡状态理论
任何形式的TST,比如微典型变分TST (microcanonical variational TST)、典型变分TST (canonical variational TST)、和改良的典型变分TST (improved canonical variational TST),其中,过渡态不一定位于马鞍点,被叫做广义的过渡态理论。
微典型变分TST
过渡态理论的一个发展,其中,对分割面进行变分处理,将针对固定的能量计算的速率最小化。在微典型变分处理下取得的速率表达式可以对能量积分,在所有能态中考虑统计分布就能得到典型变分,或热速率。
典型变分TST
典型变分TST是过渡态理论的一个发展,其中对分割面进行变分处理,在给定的温度下将速率常数最小化。
改良的典型变分TST
是对变分过渡态理论的一项改良,其中,对于域值能量以下的能量,分割面的位置取微典型变分阈值能量的位置。这样就迫使如果处于阈值能量一下的能量分布的速率常数为零。然后选择一个折衷的分割平面使具有较高能量的反应物所产生的速率常数最小化。
TST的应用: 酶反应
同样反应条件下,同一个化学反应,有没有酶催化其反应速率相差巨大。每个催化事件至少包括三步。所有的催化步骤都发生在几毫秒中,毫秒数量级的基元反应速率,是酶促反应的特点。根据过渡状态理论,催化循环的最小部分花在最重要一步,即过渡态。绝对反应速率理论定义过渡态的建议为:在反应坐标中的一个独特的、决定反应速率的物种。此后不久,莱纳斯·鲍林(Linus Pauling)指出,酶的强有力的催化作用可以用非常紧密地结合于过渡态物种来解释。 因为反应速率于在过渡态络合物中的反应物部分成正比,因此,他建议酶作用是增加反应性物种的浓度。 因为,酶典型地可以将反应速率增加至非催化反应的10-10倍,并且,Michaelis 络合物对分解反应常数的范围是:10-10 M。因而,他们提出将过渡态络合物与10-10 M范围的分解反应常数相结合。
该建议被在Chapel Hill的被卡罗林纳大学的Wolfenden极其同所完善。他们假设由酶造成的速率增高正比于酶对相对于Michaelis络合物的过渡态结构的亲和力。随着底物从Michaelis络合物朝着产物进展,在底物的电子分布方面发生酶诱导的化学过程。
酶通过质子化改变底物的电子结构、电子转移、集合扭曲、疏水分配、和与路易斯酸堿的相互作用。这些是通过依次地蛋白质和底物的构象变化而完成的。当单个的微弱的力量组合起来,集中在底物上这些单独的能量的总和导致了一个巨大的力量,能够重新定位成键电子,引起键的断裂和键的形成。很像过渡态结构的类似物因而应可作为已知的最强有力的该酶的非共价抑制剂而起到对该酶的抑制作用,甚至,如果仅仅一小部分过渡态能量该抑制剂被俘获都可明显见到抑制效应。
所有的化学转化都通过一个叫做过渡态的不稳定结构,过渡态在底物和产物的化学结构之间保持平衡。对于化学反应的过渡态人们认为有大约10 秒的寿命。在数量级上是一个单键振动的时间。没有物理的和谱学的方法可以直接观察酶催化反应的过渡态的结构,然而,过渡态的结构是了解酶催化反应的核心。因为酶的作用是降低化学反应的活化能。
现在人们普遍接收的观点是:酶的功能是稳定居于反应物和产物之间的过渡态,并且,因而,人们料想它也会于任何与该过渡态在结构上相似的该酶的抑制剂紧密结合。底物和产物经常参与几个酶催化反应,然而,过渡态只能倾向于一种参与的酶的特征,这样一种抑制剂是针对一种特定的酶的。许多过渡态抑制剂的确认支持酶催化的过渡态稳定化假设。
目前已知有大量的酶与过渡态类似物相互作用,其中大多数已经被设计用于目标酶的抑制。例子包括:HIV-1戴白酶、消旋酶类、β-内酰胺酶类、金属蛋白酶类、环氧化酶类和许多其他的酶类。
嘌呤核苷磷酸化酶
嘌呤核苷磷酸化酶(Purine nucleoside phosphorylase,P)是一个与分解代谢和核苷酸再循环有关的酶。它也是开发治疗白血病和自身免疫性疾病患者中T-细胞凋亡的新治疗药物的目标。 次黄嘌呤核苷、鸟嘌呤核苷、和2’-脱氧鸟嘌呤核苷是该酶的主要底物(图3显示一个代表性的P催化次黄嘌呤核苷底物的反应。)
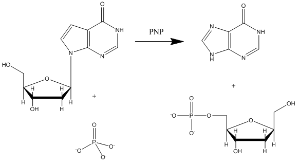
Figure 3: Purine nucleoside phosphorylase reaction with inosine substrate
阿尔伯特·爱因斯坦医学院(英语:Albert Einstein College of Medicine)(Albert Einstein College of Medicine)的 Vern Schramm(英语:Vernon L. Schramm)及其同事已经确定了P的过渡态结构并且用它巧妙地开发了紧密结合过渡态的一系列类似物用以抑制这个酶。 P的Immucillin-H(英语:Immucillin-H)抑制剂非常像公认的该过渡态结构(见图4),该抑制剂具有几处变化以使得该化合物比稍现即逝的过渡态物种更稳定。
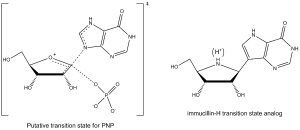
Figure 4: P transition state and immucillin-H transition state analog
P过渡态结构包含在嘌呤环N7位上用于质子化的升高的pKa。它作为氢键氢的提供者与侧链上第243位上的天门冬氨酸残基上的羰基氧形成氢键。在immucillin-H上这个位置上用9-脱氮杂嘌呤替代次黄嘌呤进行模拟。过渡态也在糖环上形成一个氧鎓离子,它由具有更稳定的核糖甙键亚氨基核糖醇部分提供。在过渡态中磷酸酯键与键的形成牵涉不多,因而,这一部分在设计类似物抑制剂时不考虑结构模拟。 过渡态类似物展示慢结合抑制物的性质,其中,第一部抑制物结合形成可逆络合物EI,然后,通过一个慢的构象改变导致一个非常稳定结合的EI*络合物。
immucillin-H结合的计量化学研究确定每个P三聚体与一个抑制剂分子相结合,而且,一个抑制剂分子就足够对这个酶产生
KI由对immucillin-H的滴定确定,并且测定它对P最初速率vo,并且该值为41 nM,KI*是由同样的速度测定计算而来的,但是用了第二稳态速率vs取代了初速率。第二稳态速率对应于当所有E已经形成EI的平衡时的稳态抑制速率。
抑制作用。此前,人们揭示P的全部催化活性每次是在分子的一个位置上进行,很像对于F1-ATP合成酶那样。人们还注意到在三个部位上全可以看到底物、产物、基态类似物的结合。因而,immucillin-H stoichiometry是抑制剂是模拟酶的过渡态的又一个线索。结构证据支持该过渡态类似物的一对三抑制作用,并且,所有基态类似物都显示完全酶占据的immucillin-H计量化学。
来自于酶过渡态分析的immucillin-H设计是开发具有药理学活性的高亲和力的酶抑制剂的强有力的方法。
参见
科廷–哈米特定律
参考文献
Laidler, K.; King, C., Development of transition-state theory. The Journal of physical chemistry 1983, 87, (15), 2657
Laidler, K., A lifetime of transition-state theory. The chemical intelligencer 1998, 4, (3), 39
Eric V. Anslyn, Dennis A. Doughtery., Transition State Theory and Related Topics. In Modern Physical Organic Chemistry University Science Books: 2006; pp 365–373
Schramm, VL., Enzymatic Transition States and Transition State Analog Design. Annual Review of Biochemistry 1998, 67, 693-720
Schramm, V.L., Enzymatic Transition State Theory and Transition State Analogue Design. Journal of Biological Chemistry 2007, 282, (39), 28297-28300
Radzicka, A.; Woldenden, R., Transition State and Multisubstrate$Analog Inhibitors. Methods in Enzymology 1995, 249, 284-312
Cleland, W.W., Isotope Effects: Determination of Enzyme Transition State Structure. Methods in Enzymology 1995, 249, 341-373
免责声明:以上内容版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。感谢每一位辛勤著写的作者,感谢每一位的分享。

相关资料





- 有价值
- 一般般
- 没价值








24小时热门







推荐阅读


关于我们

APP下载


