

更多文章
更多精彩文章




 QQ空间
QQ空间
 QQ好友
QQ好友
 微信好友
微信好友
 新浪微博
新浪微博
理论
化学键的振动是量子化的。分子会吸收特定频率的红外线,使化学键由振动基态跃迁至激发态(通常是第一激发态)。在通常状态下,分子的所有共价键几乎全部处于振动的基态。化学键的振动可用简谐振子近似,所以欲使化学键振动能级发生改变,吸收光的波数应为:
ν ν --> = 1 2 π π --> c k μ μ --> {\displaystyle \nu ={\frac {1}{2\pi c}}{\sqrt {\frac {k}{\mu }}}}
其中 π π --> {\displaystyle \pi } 为圆周率, c {\displaystyle c} 为真空中光速, k {\displaystyle k} 为化学键的“劲度系数”, μ μ --> {\displaystyle \mu } 为约化质量。约化质量由下式给出:
μ μ --> = m A m B m A + m B {\displaystyle \mu ={\frac {m_{A}m_{B}}{m_{A}+m_{B}}}}
其中 m A {\displaystyle m_{A}} 和 m B {\displaystyle m_{B}} 分别为成键原子 A {\displaystyle A} 和 B {\displaystyle B} 的质量。不同的化学键,随着成键原子的不同,约化质量也会不同;而即使对于相同的成键原子,由于化学键性质不同(比如碳碳双键和碳碳单键),其“劲度系数”也会不同。故而不同化学键会有不同的特征频率。
一个分子的总自由度为 3N ( N 为分子中原子的数量)。其中平移自由度为 3 ,分别对应对于 x 、 y 和 z 三个方向;同样的,旋转自由度亦为 3 。所以对于非线型分子,其振动模式(vibrational mode)的数量为 3N-6 。由于绕键轴旋转不计入旋转自由度,对于线型分子,振动模式的数量为 3N-5 。简单的双原子分子只有一种振动模式,那就是伸缩。更复杂的分子,其振动方式也更为复杂。例如亚甲基中的碳氢键,就可以以 “对称伸缩”、“非对称伸缩”、“剪刀式摆动”、“左右摇摆”、“上下摇摆”和“扭摆”六种方式振动(见下图)。
一般地,红外光谱上的信号数量应与分子的振动模式数量相同,但分子的振动模式若为红外活跃,必须能使分子偶极矩改变;所以并不是所有的振动模式都能在红外光谱中被观察到。此外,不同振动模式之间可以耦合,并在红外光谱上显示信号。
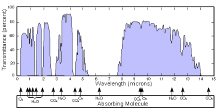
测量样品时,一束红外光穿过样品,各个波长上的能量吸收被记录下来。这可以由连续改变使用的单色波长来实现,也可以用傅立叶变换来一次测量所有的波长。这样的话,透射光谱或吸收光谱或被记录下来,显示出被样品红外吸收的波长,从而可以分析出样品中包含的化学键。
这种技术专门用在共价键的分析。如果样品的红外活跃键少、纯度高,得到的光谱会相当清晰,效果好。更加复杂的分子结构会导致更多的键吸收,从而得到复杂的光谱。但是,这项技术还是用在了非常复杂的混合物的定性研究当中。
样品制备
固态样品可溶解于有机溶剂如二氯甲烷、氯仿或甲苯中,获取光谱前先扫描相应的纯溶剂获得背景光谱,再扫描含有样品的溶液,通过计算机处理即可获得样品的红外光谱。也可将样品与矿物油混合研磨制成糊状物进行扫描。还可以将样品与溴化钾研碎混合充分混合后压片进行扫描。 液态样品可以直接通过扫描获取红外光谱,也可以制成溶液后进行扫描。
典型方法
在有机分子中吸收键的总结
运用
傅立叶变化红外光谱学
另见
红外光谱学相关表
外部键接
免责声明:以上内容版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。感谢每一位辛勤著写的作者,感谢每一位的分享。




















{{item.time}} {{item.replyListShow ? '收起' : '展开'}}评论 {{curReplyId == item.id ? '取消回复' : '回复'}}