






肌萎缩性脊髓侧索硬化症
症状和体征
由于上、下运动神经元退化导致的身体肌肉萎缩,患病者最终可能丧失发起和控制一切自主运动的能力,但膀胱、肠道和负责眼球运动的神经一般到病情晚期才会受到影响。
对于大多数患者来说其认知功能并不受影响,但少数(大约5%)仍会出现额颞叶痴呆。 大部分患者(30–50%)出现了难以察觉的认知变化,但这一变化经过详细的神经心理学测验检测出来。极少数肌萎缩侧索硬化症患者会同时出现痴呆症、退化性肌肉疾病、退行性骨疾病,这些症状都属于多系统蛋白病综合征的一部分。 感觉神经和自主神经系统一般不受影响,也就意味着大多数肌萎缩侧索硬化症患者的听觉、视觉、触觉、嗅觉和味觉都能维持正常。
早期症状
肌萎缩侧索硬化症的早期症状很难以察觉。 最早期的典型症状通常是肌肉明显的无力或/和萎缩。其他显性的症状包括吞咽困难、痉挛或牵涉的肌肉僵硬。肌无力会影响四肢;或/和出现说话含糊和带鼻音的现象。肌萎缩侧索硬化症早期出现哪些症状取决于最先受到影响的运动神经元是哪些。大约75%的患者罹患的是“四肢起病型”肌萎缩侧索硬化症,即首发症状出现在手臂和腿部。腿起病型患者会出现走路或跑步时跌到或绊倒,走路时明显拖腿而行。臂起病型患者在做一些需要灵巧手工的事时可能会遇到困难,如扣衬衫的扣子、写字或把钥匙插入锁中。少数患者这些单手或单脚的症状会维持较长的一段时间,被称为单体肌萎缩。
另外约25%的患者罹患的是“延髓起病型”肌萎缩侧索硬化症,即以说话障碍或吞咽困难为首要表现。说话开始变得含糊、带有鼻音或失语。其他症状包括吞咽困难和舌头丧失灵活性。很少一部分患者属于“呼吸起病型”肌萎缩侧索硬化症,即支持呼吸的肋间肌最先受到影响。也很少一部分患者可能伴随出现额叶痴呆症,并最终发展为其他更典型的症状。
随着病情的发展,病人开始出现运动、吞咽和构音障碍。上运动神经元受影响的症状包括肌肉僵硬和痉挛、夸张的反射(反射亢进)以及过度的呕吐反射。一个被称为“巴宾斯基征”的异常反射也是上运动神经元受损的表现。下运动神经元退化的症状包括肌肉无力和萎缩,可见的短暂的皮下肌肉抽搐(颤动)。大约15-45%的病人经历假性延髓症状——一种被称为“情绪不稳”的神经系统疾病,症状包括无法控制的大笑、哭泣或微笑。一个人只有在同时出现上下运动神经受损的症状而又没有其他的病因时,才应被诊断为肌萎缩侧索硬化症。
病程
虽然发病的顺序和程度因人而异,但是大部分病人最终都会无法行走或使用他们的双手和手臂。他们也会丧失说话和吞咽食物的能力,并最终需要依靠被称为BiPAP的便携式呼吸机。病情发展的程度可以通过《肌萎缩侧索硬化症功能评定量表修订版(ALSFRS-R)》来衡量。该测量方式是一个包含12个项目的临床访谈或自我报告的问卷调查,评分为0分(重度残疾)至48分(功能正常)。虽然有极少病人的发病过程缓慢,但是平均而言,病人每个月会丧失大约0.9的分值。一份对医师的调查表明,他们认为20%的分值减少才会有临床意义。 不管哪部分机体首先受到影响,肌肉的无力和萎缩都会随着病情的发展而蔓延到其他肢体。腿部起病的患者通常会从先发病的一只腿蔓延到另一只腿,而延髓起病的患者通常病症会先蔓延到手臂,再到腿。
40岁以下 、轻度肥胖 、并只有一肢起病、主要有上运动神经元症状. 的病人的病程会比较慢。相反,延髓起病、呼吸系统起病或前额叶痴呆起病的病人病程发展较快 。
CX3CR1等位基因突变也会影响病人病程和存活的时间。
晚期
虽然呼吸肌可以缓解呼吸问题并延长生存期,但是却不会减缓肌萎缩侧索硬化症的病程。大部分肌萎缩侧索硬化症患者从病发开始3至5年内死于呼吸衰竭。从发病到死亡的中位生存时间为39个月。只有约4%的病人存活超过10年。 吉他手杰森·贝克于1989年发病至今,而物理学家史蒂芬·霍金已经存活超过50年 , 台湾病友蔡智明于2012年发病至今仍可以靠着意志力缓慢自主行动 ,他们都是十分罕见的病例。
咀嚼和吞咽困难都会增加进食的难度,以及窒息和将食物吸入肺部的风险。在患病的晚期,吸入性肺炎会加重,而保持正常的体重也会越加困难,并有可能需要通过插管进食。随着膈肌和肋间肌的无力,呼吸也会开始衰弱。肺功能指标,比如肺活量和吸气压力都会下降。呼吸起病患者,这些症状可能在四肢发病前出现。
在患病的最后期,控制眼球运动的动眼神经以及眼外肌也会被影响。眼球运动直到最后期才受到影响很大一个原因是骨骼肌与眼外肌的差异。最终,病人的情况可能类似闭锁综合症。
眼球运动
肌萎缩侧索硬化症患者可能会难以进行自发、快速的眼球移动。眼动速度会减慢,并且平滑追踪眼动和收敛性眼动也会出现问题。 通过检测前庭眼反射可以帮助识别这些问题。眼电图(EOG)技术可以测量视网膜的静息电位。EOG结果显示患者表现出了与疾病进展相关的渐进的变化,并为疾病进展对眼部运动的影响提供了临床评价度量标准。此外,EOG也许可用于眼睛问题的早期检测。
眼外肌的胚细胞系不同于体节衍生的肌肉的胚细胞系。眼外肌很独特,因为它们在人的一生中会持续不断地重塑并保持个体随着年龄增长也能一直拥有活跃的卫星细胞。眼外肌明显有着比四肢骨骼肌更多的生肌前体细胞。
病因
遗传性
大约5%-10%的肌萎缩侧索硬化症来自家族遗传。 总而言之,第一近亲中存在一个肌萎缩侧索硬化症患者,患病的风险则增加1%。
21号染色体上的一个超氧化物歧化酶基因突变与20%家族遗传性肌萎缩侧索硬化症相关,占全部病例的2%。 这个一突变通过常染色体显性遗传方式传递的,并有一百多种不同的突变类型。引发肌萎缩侧索硬化症的最常见突变是SOD1基因突变。多见于北美患者;其特征是从发病到死亡的病程极快。在斯堪的纳维亚国家中最常见的突变是D90A-SOD1突变,其所造成的肌萎缩侧索硬化症病程较慢。且患者的存活时间平均可达11年。
2011年,一种六核苷酸重复序列的基因异常被发现于名为C9orf72的区域。这种突变被认为与肌萎缩侧索硬化症-额叶痴呆症肌萎(ALS-FTD)有联系。 6%的欧洲白人病例与此突变相关。 这个基因出现于菲律宾的后裔中。
UBQLN2基因在细胞中编码泛醌蛋白2,该蛋白是泛醌蛋白家族中的一种并能控制泛素化蛋白的降解。UBQLN2突变可阻碍蛋白质降解,导致神经退行性疾病,伴X染色体显性遗传肌萎缩侧索硬化症及肌萎缩侧索硬化症/痴呆。
一些基因突变已被证实与多种类型的肌萎缩侧索硬化症相关。已知的联系有:
SOD1 基因
在1993年,科学家们发现基因(SOD1)突变所生产的铜/锌超氧化物歧化酶(SOD1)与约20%的家族性肌萎缩性脊髓侧索硬化症相关。该酶是一种很强的抗氧化剂,能保护机体免受来源于线粒体的自由基损伤。自由基是细胞在正常代谢过程中产生的高度活性分子。自由基可积聚并导致细胞内DNA和蛋白质的损伤。到目前为止,超过110种不同的SOD1突变已被发现与代谢失调相关,其中一些(如H46R)有一个很长的临床过程;而另一些则异常迅猛,如A4V。若氧化应激防御失败,细胞则会走向凋亡。
SOD1缺陷可能是一种功能性缺失或增添。SOD1功能性缺失可能导致自由基积累。SOD1功能性增添则可能在其他方面作为毒物。
利用转基因小鼠研究SOD1在SOD1变异导致的家族性肌萎缩侧索硬化症中所扮演的角色方面已经取得了一些理论成果。SOD1基因完全缺失的小鼠通常不会罹患肌萎缩侧索硬化症,尽管会表现出与年龄有关的肌肉萎缩加速和寿命缩短。这表明SOD1变异产生的毒蛋白会导致功能的增益而不是缺失。另外,蛋白质聚集已被发现是家族性和散发性肌萎缩侧索硬化症的共同病理特征。有趣的是,存在SOD1突变的小鼠(最常见的G93A突变),突变SOD1的聚集体(错误折叠蛋白的积累)只出现于病变组织中,更多检测于运动神经元的变性过程中 。突变SOD1的总积累被怀疑会通过破坏线粒体、蛋白酶体、蛋白质折叠的分子伴侣或其他蛋白质而引起细胞功能受损 。以上任何一个环节如果被证实,将大大增加聚集体涉及突变SOD1毒性理论的可信度。批评人士指出,在人体中由SOD1突变导致的病例仅仅占所有病例的2%左右,并且发病机制可能不同于这些少数情况。到目前为止,ALS-SOD1小鼠仍然为临床前研究该疾病的最好模型,但人们也希望能开发更多有用的模型。
已存为科学界和公众提供有关肌萎缩侧索硬化症遗传学最新信息的在线数据库ALSOD。这一网站最初是在1999年为SOD1基因而开发的,但逐渐升级为包括了超过40种与肌萎缩侧索硬化症相关的基因。
其他因素
目前大约90%的病例没有家族病史,即不能明确病因。尚不确定的潜在病因包括头部外伤,兵役,频繁用药和参与身体接触性运动。
研究还集中于谷氨酸在运动神经元变性中的作用。谷氨酸是大脑中的一种神经递质。科学家发现与健康人相比肌萎缩侧索硬化症患者的血清和脑脊液中的谷氨酸含量更高 。利鲁唑是目前唯一的经FDA批准用于治疗肌萎缩侧索硬化症及目标谷氨酸运载体的药物。它对患者生存只有少量的的影响,然而研究表明过量的谷氨酸并不是导致疾病的唯一原因。
某些研究表明在少数ALS病例中,尤其是运动员,其患病与富含支链氨基酸的饮食,常见导致细胞过度兴奋的膳食补充剂之间存在联系。所提出的潜在机制是细胞过度兴奋导致了细胞对钙的吸收增加,进而导致了对钙缓冲能力极差的神经细胞死亡。
一些证据支持超氧化物歧化酶1(SOD1)蛋白在错误折叠传播上类似于朊病毒。 同样,也有人提出蓝藻毒素β-甲基-1-丙氨酸(BMAA)的结合导致了其他脘状蛋白质错误折叠的传播。
另一个非常常见的病因是运动神经元系统病变,诸如额颞叶。 这些地方的病变经常在早期就可表现出功能缺损的迹象,这一点可用来预测运动功能丧失的方向,同时这也是造成了肌萎缩侧索硬化症恶化的原因。 肌萎缩侧索硬化症的病理变化在出现任何明显的迹象或症状前就已存在。 在病程中,大约三分之一的运动神经元在肌肉萎缩变得明显之前就已死亡。
对其他一些潜在原因,包括化学品接触,电磁场暴露,职业因素,身体创伤和电击,也已进行了调查,但并未达成一致的结论。
病理生理学
肌萎缩侧索硬化症的典型特征是在大脑皮层,脑干和脊髓的上下运动神经元的死亡。在死亡之前,运动神经元在胞体和轴突中会产生富含蛋白质的包裹体。这可能是由于蛋白质降解缺陷造成的。 这些包裹体通常含有泛素,并且通常包括与肌萎缩侧索硬化症相关的蛋白中的一种,可能是SOD1,TAR DNA结合蛋白(TDP-43或TARDBP),或/和FUS。
骨骼肌运动单元
尽管共享复原的固定序列,眼外肌(EOMs)仍和骨骼肌表现出不同的特点。以下是眼外肌不同于骨骼肌运动单位的特性。
一条神经纤维只与一条或两条肌纤维连接
尽管肌梭丰富但无眼的牵张反射
无反回性抑制
无特殊的快肌或慢肌
所有的眼部运动神经元同等的参与所有类型的眼球运动——没有专门用于扫视或平滑追踪的神经元
正常的眼外肌和患者的眼外肌之间也发现存在差异。来自尸体捐赠者的眼外肌相比于四肢肌肉能保存其细胞结构。 正常的眼外肌由面向眼球的中心整体层(GL)和面向眼睑壁的薄眼睑层(OL)组成。 受肌萎缩侧索硬化症影响的眼外肌也能保存GL和OL组织。 眼外肌具有神经营养因子——脑源性神经营养因子(BDNF)和胶质细胞源性神经营养因子(GDNF),这些神经营养因子能保存受肌萎缩侧索硬化症影响的眼外肌。 层粘连蛋白是一种通常存在于神经肌肉接头(NMJ)的结构蛋白。Lnα4一种是层粘连蛋白亚型,是骨骼肌神经肌肉接头的标志。 肌萎缩侧索硬化症患者显示能保持眼外肌神经肌肉接头Lnα4的表达,但这种表达在此人的肢体肌肉神经肌肉接头中是不存在的。 层粘连蛋白的表达可能在保护肌萎缩侧索硬化症患者眼外肌的完整性中起了作用。散发性肌萎缩侧索硬化症(sALS)患者细胞内钙水平增加,导致神经递质释放的增加。散发性肌萎缩侧索硬化症患者体内血清的被动转移增加了脊髓中自发递质的释放,但不是眼外肌终端 ;因此,眼外肌被假定能耐受肌萎缩侧索硬化症导致的生理环境变化。
然而,研究者也注意到了一些现象。相比于同龄的健康者,肌萎缩侧索硬化症患者的眼外肌纤维尺寸更大。眼外肌的病变特征表现为集中和分散萎缩以及纤维肥厚,但眼外肌损伤与来自同一供体的四肢肌肉相比显著偏低。这些眼外肌还表现为在脂肪和结缔组织增生以补偿失去及萎缩的肌肉纤维。 在肌萎缩侧索硬化症患者中也出现了眼肌麻痹,在眼球运动核中以及附近的神经元损失。 此外,眼外肌纤维的肌球蛋白重链成分也发生了变化,表现为中心整体层正常表达的MyHCslow缺失,薄眼睑层不包含通常表达的MyHCemb。这种变化也许代表神经支配模式的变化,可能包括不同类型的运动神经元出现神经再支配,或缺失多个神经支配。眼外肌仅发生为MyHCslow和MyHCemb的变化,使得眼外肌纤维的成分相对正常。因为眼外肌通常是高度支配,任何去神经支配都可由邻近轴突补偿。
乳酸和肉桂酸
乳酸是糖酵解的最终产品,可导致肌肉疲劳。乳酸脱氢酶(LDH)可发挥双重功能,并能够将乳酸氧化为丙酮酸,从而被三羧酸循环利用。在眼外肌中,乳酸通路能够在活动量增加的情况下能维持肌肉收缩。因此,眼外肌中高活性的乳酸脱氢酶被认为可抵抗肌萎缩侧索硬化症。
肉桂酸是乳酸运输和摄入以耐疲劳性的外源乳酸的抑制剂。肉桂酸能够引起眼外肌疲劳,同时减少眼外肌耐力和残余力量;然而,肉桂酸对腿部的趾伸长肌没有影响。相比之下,以外源乳酸代替葡萄糖代谢增加了趾伸长肌的疲劳状况,而非减轻。 眼外肌疲劳仅仅出现于外源乳酸和肉桂酸共同取代葡萄糖代谢时。 Fatiguability in EOMs was only found when a combination of exogenous lactacte plus cinnamate replaced glucose.
诊断方法
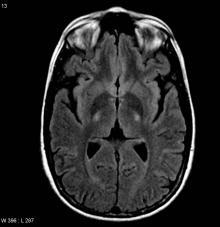
临床诊断患肌萎缩侧索硬化症者核磁共振(轴向液体衰减反转恢复序列)显示内囊后部T2信号增强
虽然单一肢体的上、下运动神经元症状强烈暗示患肌萎缩侧索硬化症的可能,但是仍没有测验能为肌萎缩侧索硬化症提供确切的诊断。相反,肌萎缩侧索硬化症的诊断主要根据医生对个体临床症状和体征的观察及一系列测试以排除其他疾病的可能。医生需要获得病人的全部病史,并通常经过定期的神经系统检测,评估如肌肉无力,肌肉萎缩,反射亢进,痉挛等症状是否恶化。
因为肌萎缩侧索硬化症的症状可能类似于大量其他更可治愈的疾病或症状,所以必须进行适当的测试以排除患其他疾病的可能。其中一项测试是肌电图(EMG),一种检测肌肉电活动的特殊记录技术。某些肌电图结果可支持肌萎缩侧索硬化症的诊断。另一项常见的测试测量神经传导速度(NCV)。但是神经传导速度结果异常也可能是由于周围神经病变(周围神经损伤)或肌病(肌肉疾病)而不是肌萎缩侧索硬化症。肌萎缩侧索硬化症常进行磁共振成像(MRI)检查,以排除脊髓肿瘤、多发性硬化症、颈部椎间盘突出、脊髓空洞症及颈椎病。
除了根据病人的症状和测试结果进行判断外,医生还可以进行血液和尿液样本检测以排除其他疾病的可能性。在某些情况下,如果医生怀疑病人可能有肌病而不是肌萎缩侧索硬化症可进行肌肉活检。
某些病毒性疾病可能会导致与肌萎缩侧索硬化症类似的症状,如人类免疫缺陷病毒(HIV)、人类T细胞白血病病毒(HTLV)、莱姆病 、梅毒 和蜱传脑炎病毒 。神经系统疾病的某些方面也与其类似,如多发性硬化症、脊髓灰质炎后综合征、多灶性运动神经病、慢性炎症性脱髓鞘性多发性神经病、脊髓性肌萎缩症和脊髓延髓肌萎缩症。
肌萎缩侧索硬化症必须与“肌萎缩侧索硬化症类似综合症”区分开,这是一种临床表现和特征与肌萎缩侧索硬化症或其变体类似的无关疾病。 由于其症状与早期肌萎缩侧索硬化症相似,所以患者应听取神经学专家的意见,从而排除一些临床可能。
但是,大多数肌萎缩侧索硬化症病例很容易诊断,大型肌萎缩侧索硬化症诊所的误诊率小于10%。 在一项研究中,190位患者符合MND / ALS的诊断标准按照研究规程和定期监测进行实验。三十例患者(16%)在临床观察发展阶段得出了与之前完全不同的诊断。 在同一项研究中,三名患者出现假阴性的诊断——重症肌无力(MG),一种自身免疫性疾病。重症肌无力症状类似于肌萎缩侧索硬化症和其他神经系统疾病,所以有时会导致延误诊断和治疗。重症肌无力是可以治疗的;肌萎缩侧索硬化症却不能。 肌无力综合征,也被称为伊顿-兰伯特综合征,类似于肌萎缩侧索硬化症并且其初期症状与肌萎缩侧索硬化症相似。
处置方法
肌萎缩侧索硬化症的处置以缓解症状与延长寿命为目的。该支持治疗最好由多学科的专业医护人员进行,尽可能的保持患者可运动及舒适。
药物治疗
利鲁唑是唯一可略微提高肌萎缩侧索硬化症患者生存率的药物。 其可延长数月生存期,并可能对延髓起病型患者延长生存期更有效。利鲁唑还可以推迟患者恶化至需要机械通气支持。服用利鲁唑的患者必须监测肝损害(发生于约10%的用药患者)。 利鲁唑由美国食品药品监督管理局批准,并由英国国家卫生与临床优化研究所推荐。但利鲁唑不能修复已有的运动神经元损伤。
其他药物可用来帮助减轻疲劳,缓解肌肉痉挛,控制痉挛状态,减少过多的唾液和痰。药物也可以帮助患者缓解疼痛,抑郁,睡眠障碍,吞咽困难,便秘等症状。巴氯芬和安定(地西泮)常被用来控制肌萎缩侧索硬化症引起的痉挛,当肌萎缩侧索硬化症患者出现吞咽口水困难时,可用苯海索或阿米替林。
呼吸支持
当协助呼吸的肌肉逐渐衰弱,可通过协助通气(间歇正压通气,双水平正压通气(BiPAP),或双相胸甲通气(BCV))辅助呼吸。这些装置通过这些直接应用于面部或身体可人为地增加患者的肺功能。当肌肉无力维持氧气和二氧化碳的水平时,这些设备就必须全天使用。双相胸甲通气可通过高频振荡及积极的呼气性呼吸协助,有增加清除呼吸道分泌物的优势。 人们最终可能会考虑机械通气的方式(呼吸器),即使用机器帮助肺收缩和膨胀的方式。为了使得效果更加理想,可能需要从鼻或口连接一个管子至气管。若要长期使用,这可考虑通过气管切开术,用一个塑料呼吸管通过颈部的开口直接插在病人的气管上。
当患者及家属决定是否及何时使用以上某一方法时,应该考虑以下几个方面。通气设备对患者生活质量的作用及所需费用因人而异。尽管呼吸支持可以缓解患者的呼吸问题并延长生存期,但它并不能阻止肌萎缩侧索硬化症病程。在作出进行通气支持的决定前,患者需充分了解这些因素以及不能移动对生活造成的长期影响,并应对生活质量问题进行深入讨论。
运用双水平正压通气的外部通气设备常被用于支持呼吸,最初用于晚上,之后也用于白天。 双水平正压通气的使用(更多时候被称为无创通气,NIV)只是一个暂时性治疗措施,在BPAP失效前,患者需决定是否进行气管切开术并长期机械通气。关于这一点,一些患者选择了姑息治疗。
物理治疗 / 职业治疗
物理治疗在肌萎缩侧索硬化症患者的病情改善上起到了很大作用。具体来说,物理及职业治疗师可以设定康复目标,并通过延长力量衰退的时间,维持耐力,减缓疼痛,预防并发症的发生,促进功能自理能力等方面提升肌萎缩侧索硬化症患者的身体素质。
职业治疗和特殊设备等辅助技术也可以提高患者在肌萎缩侧索硬化症进程中的自理和安全。温和的低强度有氧运动,如日常活动、散步、游泳、骑自行车都可以锻炼未受影响的肌肉,改善心血管健康,并可帮助患者对抗疲劳和抑郁。某些运动和伸展运动可以帮助患者预防痛苦的痉挛和肌肉的缩短(挛缩)。物理及职业治疗师可以推荐的练习,使肌肉在没有超负荷的情况下得到锻炼。他们建议使用如坡道、吊带、代步车、卫浴设备(淋浴椅,卫生间扶手等)和轮椅来帮助患者移动。职业治疗师可以提供或推荐设备并且可调整使人们在日常生活中使用时尽可能的安全和自理。
说话困难的肌萎缩侧索硬化症患者或许能够在语言病理学家那里获得帮助。这些专业保健人员可以教病人自适应策略,如一些能帮助他们说话更响亮、更清楚的方法。随着肌萎缩侧索硬化症恶化,语音病理学家会推荐使用一些强化和替代性交流的设备,如扩声器,语音发生装置(或语音输出通信设备)和/或低技术含量的通信技术,如字母板或是/否的信号。
营养治疗
肌萎缩侧索硬化症患者患者和护理人员可以向营养师学习如何计划和准备大量的小餐,保证每天可提供足够的热量,纤维和流质食物以及如何改善吞咽困难。患者可能已经开始使用吸痰器来吸除多余的液体或唾液以防止窒息。职业治疗师可以协助推荐一些适合的设备使患者自我进食更容易些。语言病理学家可推荐一些食物,以迎合他们特殊的缺陷和能力。当患者不能通过自主进食而摄取足够营养时,医生可以建议使用胃管。使用胃管可以减少因将液体吸入肺部而引起窒息和肺炎的风险。进食管不会带来疼痛,也不会妨碍病人自主进食。
研究人员表示,“肌萎缩侧索硬化症患者长期能量摄入不足,建议增加能量摄入” 并伴有严重的食欲不振 。有关动物 表明,应鼓励肌萎缩侧索硬化症患者消耗尽可能多的热量,且不要限制他们的热量摄入。截至2012年,对于体重减轻的管理依旧存在"干预措施缺乏坚实的诊据"的问题。
姑息治疗
社会工作者、家庭护理和临终关怀护士会帮助肌萎缩侧索硬化症患者,他们的家人和照顾者应对医疗、情感、经济方面的问题,特别是在疾病的晚期。社会工作者可提供,如经济援助、安排律师,准备遗嘱、为病人和医护人员的寻找支持团体等方面提供支持。家庭护士不仅可以提供医疗保健,而且会教导照顾者如何使用呼吸器、喂食,及在移动病人时避免皮肤疼痛问题和挛缩。家庭临终关怀护士会咨询医生,以确保适当的药物治疗,控制疼痛,和其他会提高希望留在家里的患者的生活质量。家庭临终关怀团队也对病人及照顾者就临终问题提供专业咨询。
流行病学
在世界上许多地区,肌萎缩侧索硬化症的发病率都还是未知的。 在欧洲,每年每十万人中约有2.2人确诊。 在美国,每年有超过5600人确诊,有超过3万患者。肌萎缩侧索硬化症每年约导致十万中2人死亡。
虽然肌萎缩侧索硬化症被归入罕见疾病,但其却是最常见的运动神经元疾病。所有种族及民族背景都可能患病。每年每十万人中就有一到二人罹患肌萎缩性侧索硬化症。 据估计每十万白种人中有1.2至4.0人罹患肌萎缩性侧索硬化症,其他种族患病率则较低。 菲律宾人肌萎缩性侧索硬化症发病率仅次于白人,为每十万人中有1.1至2.8人患病。
有一些关于“集群”患病的报道,包括旧金山49人队的三名橄榄球运动员 ,意大利的超过50个足协球员 ,英国南部的足协球员的三个朋友,以及法国南部的一对配偶(丈夫和妻子)。 尽管一些人认为肌萎缩性侧索硬化症是基因和环境共同作用的结果,但除了随年龄增长风险更高外,仍未确认环境对该病的影响。
历史
肌萎缩性侧索硬化症至少可以追溯至1824年查尔斯·贝尔的描述。
英国科学家奥古斯都·沃勒在1850年描述了萎缩神经纤维的外观。1869年,法国医生让-马丁·沙可首次将这些症状与神经系统病变联系在一起,并在1874年发表论文以肌萎缩性侧索硬化症为名介绍此病。 1881年,该论文被翻译成了英语,出版在《神经系统疾病讲座》( Lectures on the Diseases of the Nervous System )第三卷。
美国传奇棒球运动员卢·贾里格在1939年发生罹患此病,于7月4日发表了告别演说,肌萎缩性侧索硬化症就此在美国而闻名。两年后卢·贾里格即离世。 2014年,因冰桶挑战而在全世界闻名。
在20世纪50年代,一次肌萎缩性侧索硬化症疫情于关岛的查莫罗人中爆发。
1991年,研究者发现了21号染色体与家族性肌萎缩性侧索硬化症(FALS)联系在一起。1993年,发现21号染色体的SOD1基因在某些家族性肌萎缩性侧索硬化症病例中发挥的作用。1996年,利鲁唑成为第一个美国食品药品监督局批准用于肌萎缩性侧索硬化症的药物。
1998年,埃斯科里亚尔标准(El Escorial)被确定为肌萎缩性侧索硬化症患者临床研究分类的标准。第二年,修订后的肌萎缩性侧索硬化症功能评分量表发表,并很快成为肌萎缩性侧索硬化症患者临床研究恶化评级的黄金标准。2011年,发现C9ORF72中非编码的重复序列扩增是导致肌萎缩性侧索硬化症和额颞叶痴呆的主要原因。
词源
肌萎缩性侧索硬化症的学名来源于希腊文“amyotrophia”:其中“a-”表示“不”,“myo”指“肌肉”,trophia意思是“营养”;因此“amyotrophia”表示“没有肌肉营养”,描述了患者肌肉组织的萎缩特征。脊髓横向识别领域是受影响的神经细胞所在部位。在这一地区的退化会导致疤痕或硬化。
延伸阅读
ALS Hope Foundation. [ 2008-06-21 ] . (原始内容存档于12 May 2008). Dedicated to the care and cure of people with Lou Gehrig"s Disease. (from site page us.php)
Lou Gehrig: The Official Web Site. CMG Worldwide. [ 2008-06-21 ] . (原始内容存档于9 May 2008). The Official Web site of Lou Gehrig is an informational Web site intended to honor the life, the legend and the career of Lou Gehrig. (from site page siteinfo/index.htm)
Patrick Aebischer; Ann C. Kato.Playing defense against Lou Gehrig"s Disease (Paper) . Scientific American (Verlagsgruppe Georg von Holtzbrinck). November 2007: 86–93 [ 2008-06-21 ] . Researchers have proposed potential therapies for a paralyzing disorder once thought to be untreatable (sub-title)
免责声明:以上内容版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。感谢每一位辛勤著写的作者,感谢每一位的分享。

相关资料




- 有价值
- 一般般
- 没价值















推荐阅读




关于我们

APP下载


