






苯
发现
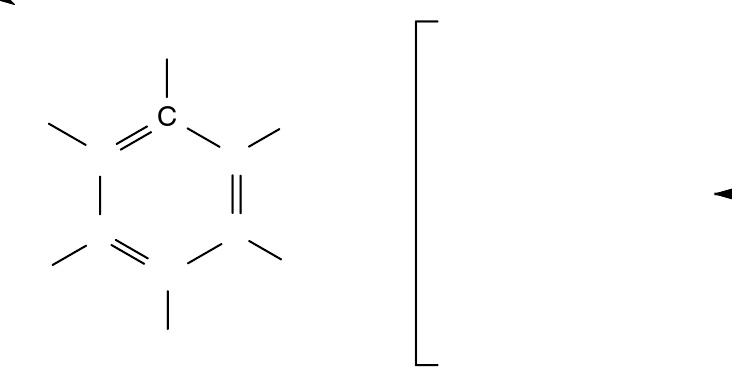
凯库勒的摆动双键
苯最早是在18世纪初研究将煤气作为照明用气时合成出来的。一般认为苯是在1825年由麦可·法拉第发现的。他从鱼油等类似物质的热裂解产品中分离出了较高纯度的苯,称之为“氢的重碳化物”( Bicarburet of hydrogen )。 并且测定了苯的一些物理性质和它的化学组成,阐述了苯分子的碳氢比。
1833年, 米修里希·伊尔哈得 ( 英语 : Eilhard Mitscherlich ) 确定了苯分子中6个碳和6个氢原子的经验式(C 6 H 6 )。 弗里德里希·凯库勒于1865年提出了苯环单、双键交替排列、无限共轭的结构,即现在所谓“凯库勒式”。据称他是因为梦到一条蛇咬住了自己的尾巴才受到启发想出“凯库勒式”的。 他又对这一结构作出解释说环中双键位置不是固定的,可以迅速移动,所以造成6个碳等价。他通过对苯的一氯代物、二氯代物种类的研究,发现苯是环形结构,每个碳连接一个氢。也有说法指出,把苯的分子结构画成六角形环状结构最早是法国化学家奥古斯特·劳伦1854年在《化学方法》一书中提出的。但是出于某种原因,凯库勒在论文没有提及劳伦的成果。
也有人提出了其他的设想:比如詹姆斯·杜瓦就归纳出不同结构;以其命名的杜瓦苯现已被证实是与苯不同的另外一种物质,可由苯经光照得到。
1845年德国化学家霍夫曼从煤焦油的轻馏分中发现了苯,他的学生查尔斯·曼斯菲尔德(Charles Mansfield)随后进行了加工提纯。后来他又发明了结晶法精制苯。他还进行工业应用的研究,开创了苯的加工利用途径。大约从1865年起开始了苯的工业生产。最初是从煤焦油中回收。随着它的用途的扩大,产量不断上升,到1930年已经成为世界十大吨位产品之一。
结构
苯具有的苯环结构导致它有特殊的芳香性。苯环是最简单的芳环,由六个碳原子构成一个六元环,每个碳原子接一个基团,苯的6个基团都是氢原子。
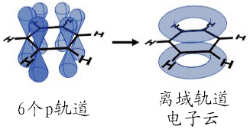
6个p轨道形成离域大Π键的电子云
苯不是单、双键交替排列的轮烯,原子间成键并不是不连续的单双键交替,而是被离域π电子云覆盖。参见芳香环。
苯分子是平面分子,12个原子处于同一平面上,6个碳和6个氢是均等的,C-H键长为1.08Å,C-C键长为1.40Å,此数值介于单双键长之间。分子中所有键角均为120°,说明碳原子都采取sp 杂化。这样每个碳原子还剩余一个p轨道垂直于分子平面,每个轨道上有一个电子。于是6个轨道重叠形成离域大Π键(即π 6 ),存在下图所示的共振式,现在认为这是苯环非常稳定的原因,也直接导致了苯环的芳香性。
从分子轨道理论来看,可以认为苯的6个 p 轨道相互作用形成6个π分子轨道,其中ψ 1 、ψ 2 、ψ 3 是能量较低的成键轨道,ψ 4 、ψ 5 、ψ 6 是能量较高的反键轨道。ψ 2 、ψ 3 和ψ 4 、ψ 5 是两对简并轨道。基态时苯的电子云分布是三个成键轨道叠加的结果,故电子云均匀分布于苯环上下及环原子上,形成闭合的电子云。它是苯分子在磁场中产生环电流的根源。
物理性质
苯的沸点为80.1℃,熔点为5.5℃,在常温下是一种无色、有芳香气味的透明液体,易挥发。苯比水密度低,密度为0.88g/ml,但其分子质量比水重。苯难溶于水,1升水中最多溶解1.8g苯;但苯是一种良好的有机溶剂,溶解有机分子和一些非极性的无机分子的能力很强。
苯能与水生成恒沸物,沸点为69.25℃,含苯91.2%。因此,在有水生成的反应中常加苯蒸馏,以将水带出。
在10~1500毫米Hg之间的饱和蒸气压可以根据安托万方程计算:
其中:P单位为mmHg,t单位为℃,A = 6.91210,B = 1214.645,C = 221.205。
化学性质
苯参加的化学反应大致有3种:一种是其他基团和苯环上的氢原子之间发生的取代反应;一种是发生在C-C双键上的加成反应;一种是苯环的断裂。
取代反应
苯环上的氢原子在一定条件下可以被卤素、硝基、磺酸基、烃基等取代,生成相应的衍生物。由于取代基的不同以及氢原子位置的不同、数量不同,可以生成不同数量和结构的同分异构体。
苯环的电子云密度较大,所以发生在苯环上的取代反应大都是亲电取代反应。亲电取代反应是芳环有代表性的反应。苯的取代物在进行亲电取代时,第二个取代基的位置与原先取代基的种类有关。
卤代反应
苯的卤代反应的通式可以写成:
反应过程中,卤素分子在苯和催化剂的共同作用下异裂,X 进攻苯环,X 与催化剂结合。以溴为例:反应需要加入铁粉,铁在溴作用下先生成三溴化铁。
反应机理为:
在工业上,卤代苯中以氯和溴的取代物最为重要。
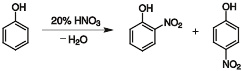
硝化反应
苯和硝酸在浓硫酸作催化剂的条件下可生成硝基苯:
硝化反应是一个强烈的放热反应,很容易生成一取代物,但是进一步反应速度较慢。
磺化反应
用浓硫酸或者发烟硫酸在较高温度下可以将苯磺化成苯磺酸。
磺化是可逆反应,苯磺酸在加热下与稀硫酸或盐酸反应,可失去磺基,生成苯。所以在合成时可以通过磺化反应保护芳核上的某一位置。
苯环上引入一个磺酸基后反应能力下降,不易进一步磺化,需要更高的温度才能引入第二、第三个磺酸基。这说明硝基、磺酸基都是钝化基团,即妨碍再次亲电取代进行的基团。
傅-克反应
在AlCl 3 催化下,苯也可以和醇、烯烃和卤代烃反应,苯环上的氢原子被烷基取代生成烷基苯。这种反应称为烷基化反应,又称为傅-克烷基化反应。 例如与乙烯烷基化生成乙苯:
在反应过程中,R基可能会发生重排:如1-氯丙烷与苯反应生成异丙苯,这是由于自由基总是趋向稳定的构型。
在强路易斯酸催化下,苯与酰氯或者羧酸酐反应,苯环上的氢原子被酰基取代生成酰基苯。反应条件类似烷基化反应。
加成反应
苯环虽然很稳定,但是在一定条件下也能够发生双键的加成反应。通常经过催化加氢,兰尼镍/钯/铂作催化剂,苯可以生成环己烷。
此外由苯生成六氯环己烷(六六六)的反应可以在紫外线照射的条件下,由苯和氯气加成而得。
氧化反应
苯和其他的烃一样,都能燃烧。当氧气充足时,产物为二氧化碳和水。
苯在特定情况下也可被臭氧氧化,产物是乙二醛。这个反应可以看作是苯的离域电子定域后生成的环状多烯烃发生的臭氧化反应。
但是在一般条件下,苯不能被强氧化剂所氧化。但是在氧化钼、五氧化二钒等催化剂存在下,与空气中的氧在500℃时反应,苯可以选择性的氧化成顺丁烯二酸酐。这是屈指可数的几种能破坏苯的六元碳环系的反应之一。(马来酸酐是五元杂环。)
这是一个强烈的放热反应。
其他反应
苯在高温下,用铁、铜、镍做催化剂,可以发生缩合反应生成联苯。 和甲醛及次氯酸在氯化锌存在下可生成氯甲基苯。和乙基钠等烷基金属化物反应可生成苯基金属化物。在四氢呋喃中氯苯或溴苯和镁反应可生成苯基格林尼亚试剂。
苯环的亲电取代定位效应
第一类邻、对位定位基
邻、对位定位基亦被称为 o (ortho)、 p (para)定位基,可使新的取代基主要进入邻、对位( o + p >60%),除了卤素以外都可使苯环活化,有利于亲电取代反应的进行。
第二类间位定位基
间位定位基亦被称为 m (meta)定位基,可使新的定位基主要进入间位( m >40%),并使苯环钝化,不利于亲电取代反应的进行。
制备
苯可以由含碳量高的物质不完全燃烧获得。自然界中,火山爆发和森林火险都能生成苯。苯也存在于香烟的烟中。
直至二战,苯还是一种钢铁工业焦化过程中的副产物。这种方法只能从1吨煤中提取出1千克苯。1950年代后,随着工业上,尤其是日益发展的塑料工业对苯的需求增多,由石油生产苯的过程应运而生。现在全球大部分的苯来源于石油化工。工业上生产苯最重要的三种过程是催化重整、甲苯加氢脱烷基化和蒸汽裂化。
从煤焦油中提取
在煤炼焦过程中生成的轻焦油含有大量的苯。这是最初生产苯的方法。将生成的煤焦油和煤气一起通过洗涤和吸收设备,用高沸点的煤焦油作为洗涤和吸收剂回收煤气中的煤焦油,蒸馏后得到粗苯和其他高沸点馏分。粗苯经过精制可得到工业级苯。这种方法得到的苯纯度比较低,而且环境污染严重,工艺比较落后。
从石油中提取
在原油中含有少量的苯,从石油产品中提取苯是最广泛使用的制备方法。
催化重整
重整这里指使脂肪烃成环、脱氢形成芳香烃的过程。这是从第二次世界大战期间发展形成的工艺。
在500-525 ℃、8-50个大气压下,各种沸点在60-200 ℃之间的脂肪烃,经铂-铼催化剂,通过脱氢、环化转化为苯和其他芳香烃。从混合物中萃取出芳香烃产物后,再经蒸馏即分出苯。也可以将这些馏分用作高辛烷值汽油。
蒸汽裂解
蒸汽裂解是由乙烷、丙烷或丁烷等低分子烷烃以及石脑油、重柴油等石油组份生产烯烃的一种过程。其副产物之一裂解汽油富含苯,可以分馏出苯及其他各种成分。裂解汽油也可以与其他烃类混合作为汽油的添加剂。
裂解汽油中苯大约有40-60%,同时还含有二烯烃以及苯乙烯等其他不饱和组份,这些杂质在贮存过程中易进一步反应生成高分子胶质。所以要先经过加氢处理过程来除去裂解汽油中的这些杂质和硫化物,然后再进行适当的分离得到苯产品。
芳烃分离
从不同方法得到的含苯馏分,其组分非常复杂,用普通的分离方法很难见效,一般采用溶剂进行液-液萃取或者萃取蒸馏的方法进行芳烃分离,然后再采用一般的分离方法分离苯、甲苯、二甲苯。根据采用的溶剂和技术的不同又有多种分离方法。
Udex法:由美国道化学公司和UOP公司在1950年联合开发,最初用二乙二醇醚作溶剂,后来改进为三乙二醇醚和四乙二醇醚作溶剂,过程采用多段升液通道(multouocomer)萃取器。苯的收率为100%。
Suifolane法:荷兰壳牌公司开发,专利为UOP公司所有。溶剂采用环丁砜,使用转盘萃取塔进行萃取,产品需经白土处理。苯的收率为99.9%。
Arosolvan法:由联邦德国的鲁奇公司在1962年开发。溶剂为N-甲基吡咯烷酮(NMP),为了提高收率,有时还加入10-20%的乙二醇醚。采用特殊设计的Mechnes萃取器,苯的收率为99.9%。
IFP法:由法国石油化学研究院在1967年开发。采用不含水的二甲亚砜作溶剂,并用丁烷进行反萃取,过程采用转盘塔。苯的收率为99.9%。
Formex法:为意大利SNAM公司和LRSR石油加工部在1971年开发。吗啉或N-甲酰吗啉作溶剂,采用转盘塔。芳烃总收率98.8%,其中苯的收率为100%。
甲苯脱烷基化
甲苯脱烷基制备苯,可以采用催化加氢脱烷基化,或是不用催化剂的热脱烷基。原料可以用甲苯、及其和二甲苯的混合物,或者含有苯及其他烷基芳烃和非芳烃的馏分。
甲苯催化加氢脱烷基化
用铬、钼或氧化铂等作催化剂,500-600 ℃高温和40-60个大气压的条件下,甲苯与氢气混合可以生成苯,这一过程称为加氢脱烷基化作用。如果温度更高,则可以省去催化剂。反应按照以下方程式进行:
根据所用催化剂和工艺条件的不同又有多种工艺方法:
Hydeal法:由Ashiand & refing和UOP公司在1961年开发。原料可以是重整油、加氢裂解汽油、甲苯、碳6-碳8混合芳烃、脱烷基煤焦油等。催化剂为氧化铝-氧化铬,反应温度600-650℃,压力3.43-3.92MPa。苯的理论收率为98%,纯度可达99.98%以上,质量优于Udex法生产的苯。
Detol法:Houdry公司开发。用氧化铝和氧化镁做催化剂,反应温度540-650℃,反应压力0.69-5.4MPa,原料主要是碳7-碳9芳烃。苯的理论收率为97%,纯度可达99.97%。
Pyrotol法:Air products and chemicals公司和Houdry公司开发。适用于从乙烯副产裂解汽油中制苯。催化剂为氧化铝-氧化铬,反应温度600-650℃,压力0.49-5.4MPa。
Bextol法:壳牌公司开发。
BASF法:BASF公司开发。
Unidak法:UOP公司开发。
甲苯热脱烷基化
甲苯在高温氢气流下可以不用催化剂进行脱烷基制取苯。反应为放热反应,针对遇到的不同问题,开发出了多种工艺过程。
MHC加氢脱烷基过程:由日本三菱石油化学公司和千代田建设公司在1967年开发。原料可以用甲苯等纯烷基苯,含非芳烃30%以内的芳烃馏分。操作温度500-800℃,操作压力0.98MPa,氢/烃比为1-10。过程选择性97-99%(mol),产品纯度99.99%。
HDA加氢脱烷基过程:由美国Hydrocarbon Research和Atlantic Richfield公司在1962年开发。原料采用甲苯,二甲苯,加氢裂解汽油,重整油。从反应器不同部位同如氢气控制反应温度,反应温度600-760℃,压力3.43-6.85MPa,氢/烃比为1-5,停留时间5-30秒。选择性95%,收率96-100%。
Sun过程:由Sun Oil公司开发
THD过程:Gulf Research and Development公司开发
Monsanto过程:孟山都公司开发
甲苯歧化和烷基转移
随着二甲苯用量的上升,在1960年代末相继开发出了可以同时增产二甲苯的甲苯歧化和烷基转移技术,主要反应为:
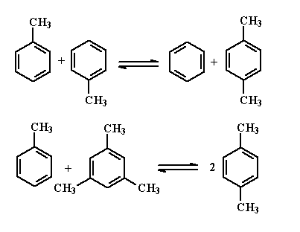
这个反应为可逆反应,根据使用催化剂、工艺条件、原料的不同而有不同的工艺过程。
LTD液相甲苯岐化过程:美国美孚化学公司在1971年开发,使用非金属沸石或分子筛催化剂,反应温度260-315℃,反应器采用液相绝热固定床,原料为甲苯,转化率99%以上
Tatoray过程:日本东丽公司和UOP公司1969年开发,以甲苯和混合碳9芳烃为原料,催化剂为丝光沸石, 反应温度350-530℃,压力2.94MPa,氢/烃比5-12,采用绝热固定床反应器,单程转化率40%以上,收率95%以上,选择性90%,产品为苯和二甲苯混合物。
Xylene plas过程:由美国Atlantic Richfield公司和Engelhard公司开发。使用稀土Y型分子筛做催化剂,反应器为气相移动床,反应温度471-491℃,常压。
TOLD过程:日本三菱瓦斯化学公司1968年开发,氢氟酸-氟化硼催化剂,反应温度60-120℃,低压液相。有一定腐蚀性。
其他方法
此外,苯还可以通过乙炔三聚得到,但产率很低。具体的做法是,将乙炔通过高温石英管(500℃)。反应方程式如下:
此反应实则为狄尔斯-阿尔德反应。
分析测试方法
气相色谱和液相色谱可以检测各种产品中苯的含量。苯的纯度的测定一般使用冰点法。
对空气中微量苯的检测,可以用甲基硅油等有挥发性的有机溶剂或者低分子量的聚合物吸收,然后通过色谱进行分析;或者采用比色法分析;也可以将含有苯的空气深度冷冻,将苯冷冻下来,然后把硫酸铁和过氧化氢溶液加入得到黄褐色或黑色沉淀,再用硝酸溶解,然后通过比色法分析。或者直接用硝酸吸收空气中的苯,硝化成间二硝基苯,然后用二氯化钛溶液滴定,或者用间二甲苯配制的甲乙酮碱溶液比色定量。
安全
毒性
由于苯的挥发性大,暴露于空气中很容易扩散。人和动物吸入或皮肤接触大量苯进入体内,会引起急性和慢性苯中毒。有研究报告表明,引起苯中毒的部分原因是由于在体内苯生成了苯酚。
苯对中枢神经系统产生麻痹作用,引起急性中毒。重者会出现头痛、恶心、呕吐、神志模糊、知觉丧失、昏迷、抽搐等,严重者会因为中枢系统麻痹而死亡。少量苯也能使人产生睡意、头昏、心率加快、头痛、颤抖、意识混乱、神志不清等现象。摄入含苯过多的食物会导致呕吐、胃痛、头昏、失眠、抽搐、心率加快等症状,甚至死亡。吸入20000ppm的苯蒸气5-10分钟便会有致命危险。
长期接触苯会对血液造成极大伤害,引起慢性中毒,引起神经衰弱综合症。苯可以损害骨髓,使红血球、白细胞、血小板数量减少,并使染色体畸变,从而导致白血病,甚至出现再生障碍性贫血。苯可以导致大量出血,从而抑制免疫系统的功用,使疾病有机可乘。有研究报告指出,苯在体内的潜伏期可长达12-15年。
妇女吸入过量苯后,会导致月经不调达数月,卵巢会缩小。苯对胎儿发育和对男性生殖力的影响,尚未明了。孕期动物吸入苯后,会导致幼体的重量不足、骨骼延迟发育、骨髓损害。
苯对皮肤、粘膜有刺激作用。国际癌症研究中心(IARC)已经确认苯为致癌物。
接触限值:
中国TWA 6 mg/m (皮),STEL 10 mg/m
美国ACGIH 10ppm, 32 mg/m TWA: OSHA 1ppm, 3.2 mg/m
毒性:
LD50: 930 mg/kg(大鼠经口);48 mg/kg(小鼠经皮)
LC50: 10000ppm 7小时(大鼠吸入)
当然,由于每个人的健康状况和接触条件不同,对苯的敏感程度也不相同。嗅出苯的气味时,它的浓度大概是1.5ppm,这时就应该注意到中毒的危险。在检查时,通过尿和血液的检查可以很容易查出苯的中毒程度。
代谢
苯主要通过呼吸道吸入(47-80%)、胃肠及皮肤吸收的方式进入人体。一部分苯可通过尿液排出,未排出的苯则首先在肝中细胞色素P450单加氧酶作用下被氧分子氧化为环氧苯(7-氧杂双环[4.1.0]庚-2,4-二烯)。环氧苯与它的重排产物氧杂环庚三烯存在平衡,是苯代谢过程中产生的有毒中间体。接下来有三种代谢途径:与谷胱甘肽结合生成苯巯基尿酸;继续代谢为苯酚、邻苯二酚、对苯二酚、偏苯三酚、邻苯醌、对苯醌等,以葡萄糖苷酸或硫酸盐结合物形式排出;以及被氧化为已二烯二酸。
乙醇和甲苯可以降低苯的毒性。
可燃性
由于苯可以在空气中燃烧,因此它一般都被定为危险品。例如在中华人民共和国国家标准《危险货物品名表》(GB 12268-90)中,苯属第三类危险货物易燃液体中的中闪点液体。 而且由于它的挥发性,可能造成蒸气局部聚集,因此在贮存、运输时一般都要求远离火源和热源,防止静电。
由于苯的冰点比较高(5.5℃),在寒冷天气中运输会有困难,但是加热熔化会带来危险性。
工业用途
早在1920年代,苯就已是工业上一种常用的溶剂,主要用于金属脱脂。由于苯有毒,人体能直接接触溶剂的生产过程现已不用苯作溶剂。
苯有减轻爆震的作用而能作为汽油添加剂。在1950年代四乙基铅开始使用以前,所有的抗爆剂都是苯。然而现在随着含铅汽油的淡出,苯又被重新起用。由于苯对人体有不利影响,对地下水质也有污染,欧美国家限定汽油中苯的含量不得超过1%。至2011年时,美国国家环境保护局将进一步收紧限制,令汽油的苯含量上限进一步降低至0.62%。
苯在工业上最重要的用途是做化工原料。苯可以合成一系列苯的衍生物:
苯与乙烯生成乙苯,后者可以用来生产制塑料的苯乙烯;
与丙烯生成异丙苯,后者可以经异丙苯法来生产丙酮与制树脂和粘合剂的苯酚;
制尼龙的环己烷;
合成顺丁烯二酸酐;
用于制作苯胺的硝基苯;
多用于农药的各种氯苯;
合成用于生产洗涤剂和添加剂的各种烷基苯。
此外还可以用来合成氢醌,蒽醌等化工产品。
食品中的含量
在若干软性饮品,如部分汽水、果汁饮品、果汁味饮品、葡萄适中,其中的苯甲酸盐类防腐剂(包括苯甲酸钠、苯甲酸钾、苯甲酸钙) 会跟维他命C(抗坏血酸)或异抗坏血酸发生化学作用,脱羧,形成少量的致癌物质苯。该反应在光照或加热时加速进行。多数饮料中,苯的含量都在10μg/kg以下,符合世界卫生组织的标准(10ppb),但高于美国(5ppb) 、加拿大(5ppb)和欧盟(1ppb) 的标准。有少量饮料含有较高含量的苯,含量最高的可达87.9ppb,但与日常生活中吸入的苯含量相比,这个数值仍然较小。例如,人每天吸入的洁净空气中约含苯220μg;人开车一小时会吸入40μg的苯;每天吸20支烟的人约会吸入7900μg的苯(欧盟估计值),通过被动吸烟吸入的苯也有63μg。
2008年,可口可乐公司宣布,将逐渐在其饮料中(除芬达和乐倍外)禁止苯甲酸钠的使用。
苯的异构体
杜瓦苯
休克尔苯(盆苯)
棱柱烷
苯的衍生物
下面是一些有代表性的苯的取代物或与苯结构相似的物质。
取代苯
烃基取代:甲苯、二甲苯、苯乙烯
含氧基团取代:苯酚、苯甲酸、苯乙酮、苯醌(对苯醌、邻苯醌)
卤代:氯苯、溴苯
多环芳烃
联苯、三联苯
稠环芳烃:萘、蒽、菲、茚、芴、苊、薁、富勒烯
参见
芳香性
粗苯
参考文献
书籍
魏文德主编,《有机化工原料大全》第三卷,化学工业出版社,1994年,p358-381,ISBN 978-7-5025-0684-1
[英]汉考克(Hancock, E.G.)主编,《苯及其工业衍生物》,化学工业出版社,1982.11
Wilson, L. D. "Health Hazards from aromatic Hydrocarbons", Des Plaines, III., Universal Oil Products Company, 1962
尹冬冬主编,《有机化学》上册,高等教育出版社,2003年
其他
中国石化北京化工研究院,《常用危险化学品安全数据卡》(内部材料),2004年
免责声明:以上内容版权归原作者所有,如有侵犯您的原创版权请告知,我们将尽快删除相关内容。感谢每一位辛勤著写的作者,感谢每一位的分享。




- 有价值
- 一般般
- 没价值
















推荐阅读

关于我们

APP下载


